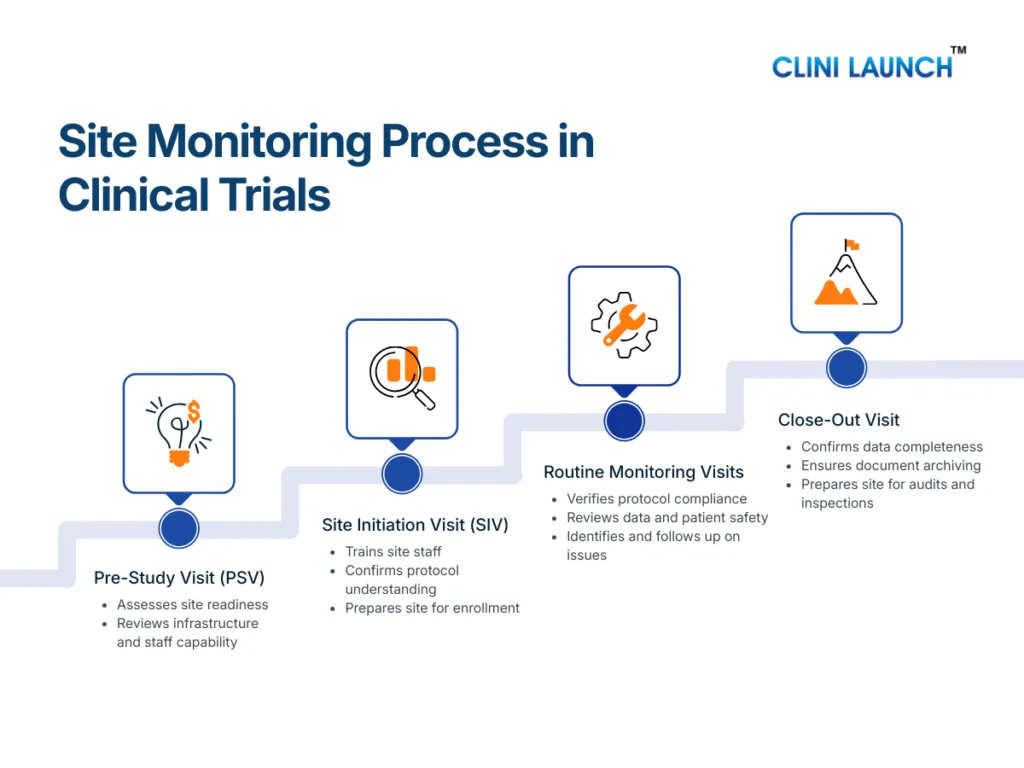
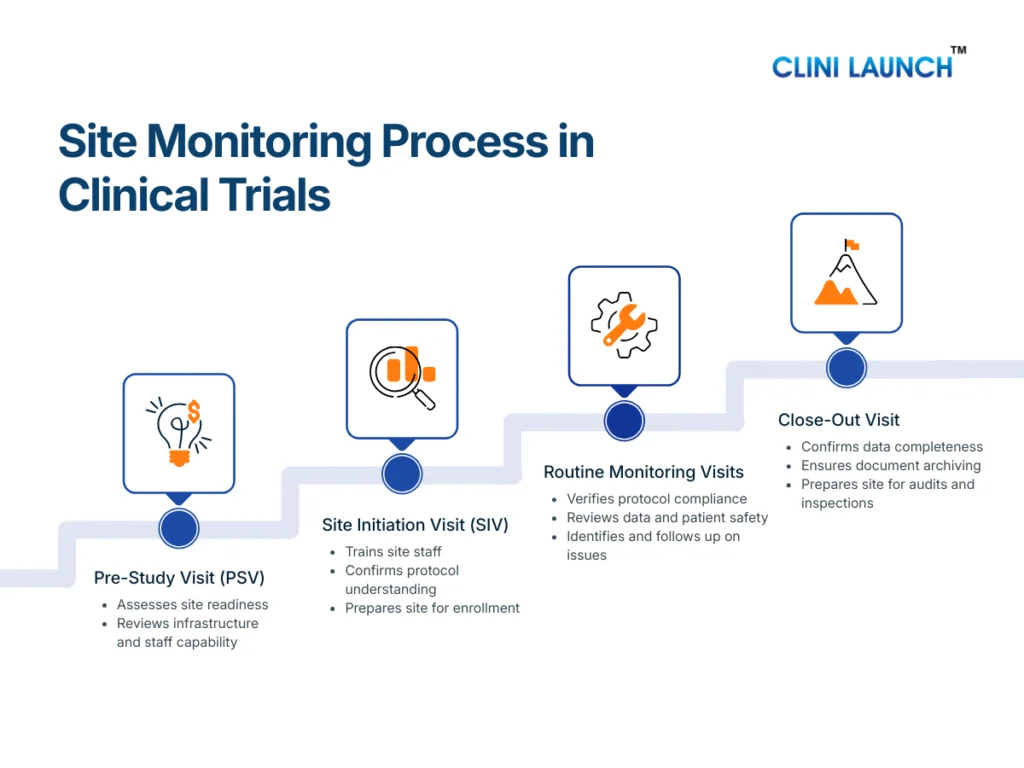
Medical coder roles and responsibilities vary by care setting. Inpatient coding covers full hospital stays, outpatient coding focuses on clinic visits and same-day services, emergency department coding handles urgent care encounters, and ancillary coding supports diagnostic and technical services. Each role differs in documentation depth, decision pressure, and compliance risk.
Medical coding is often misunderstood as a single, routine job where professionals simply convert diagnoses and procedures into codes. In reality, medical coder roles and responsibilities vary significantly depending on where patient care takes place and the type of services delivered. Coding a full hospital admission is fundamentally different from coding a clinic visit, an emergency case, or a diagnostic service, and each carries its own level of complexity, accountability, and regulatory scrutiny.
In real healthcare environments, medical coders play a critical role in ensuring accurate documentation, compliant billing, and smooth functioning of the healthcare revenue cycle. Their work directly affects reimbursement, audit outcomes, and the quality of healthcare data used for reporting and decision-making. Understanding how medical coding responsibilities change across inpatient, outpatient, emergency department, and ancillary settings is essential for anyone exploring medical coding as a career or trying to build realistic job readiness.
This article breaks down who a medical coder is, the different types of medical coding roles that exist, the specific responsibilities attached to each role, and how these responsibilities translate into real day-to-day work inside healthcare organizations.
Who Is a Medical Coder?
A medical coder is a healthcare professional who reviews clinical documentation and converts it into standardized medical codes used for billing, compliance, and reimbursement. Their work directly affects claim accuracy, audit outcomes, and the healthcare revenue cycle.
The duties of a medical coder include interpreting physician notes, discharge summaries, procedure reports, and diagnostic findings, then applying the correct codes based on official guidelines. This role requires accuracy and judgment, not data entry, because coding errors can lead to denials, compliance issues, and revenue loss.
Medical coding is not a single role. Medical coder roles and responsibilities vary depending on where patient care takes place. Based on the care setting, medical coders typically work in:
Inpatient (IP) medical coding – Hospital admissions
Outpatient (OP) medical coding – Clinic visits and same-day services
Emergency department (ED) coding – Urgent and unplanned care
Ancillary medical coding – Diagnostic and technical services
Each role differs in complexity, documentation depth, and work pace. Understanding these differences is essential before learning what responsibilities each role actually carries.
Choosing the Right Medical Coding Role
Aspect
Inpatient (IP)
Outpatient (OP)
Emergency Department (ED)
Ancillary Services
Work pace
Slower and methodical
Steady and consistent
Fast and time-sensitive
Steady and task-focused
Complexity level
High
Moderate
Moderate to high
Moderate
Documentation depth
Very detailed (entire hospital stay)
Moderate (single visit or service)
Brief and evolving
Structured and report-based
Decision intensity
High (multiple diagnoses and procedures)
Moderate (rule-driven decisions)
High (quick judgment required)
Moderate (precision-based decisions)
Entry vs advanced suitability
Better suited after experience
Common entry point for beginners
Entry to intermediate (with guidance)
Entry to intermediate (with structured training)
Who this role fits best
Detail-oriented, analytical learners
Learners who prefer structure
Those comfortable with pressure
Coders who like precision and technical work
Advanced Diploma in
Medical Coding
Build practical skills in medical coding and clinical documentation used across hospitals, healthcare providers, and global healthcare services. Learn how diagnoses, procedures, and services are translated into standardized medical codes while ensuring accuracy, compliance, and reimbursement integrity.
Medical coder roles and responsibilities are defined by where care is delivered, not by job titles. Each role carries different decision pressure, documentation depth, and impact on the healthcare revenue cycle. Treating them as the same role is how beginners get blindsided later.
1.Inpatient Medical Coding (IP)
Inpatient medical coding applies when a patient is formally admitted to the hospital, usually for 24 hours or more. Unlike other roles, inpatient coding covers the entire hospital stay, from admission through discharge.
The coder is responsible for translating a complex clinical journey into codes that accurately reflect:
Why the patient was admitted
What conditions were treated
What procedures were performed
How severe the case was
This role relies heavily on ICD 10 CM coding for diagnoses and ICD-10-PCS for inpatient procedures.
Core Responsibilities
Review the full medical record from admission to discharge
Identify and assign the principal diagnosis
Code all relevant secondary diagnoses, including complications and comorbidities
Apply proper sequencing rules to reflect severity of illness
Code inpatient procedures accurately
Support correct DRG assignment and compliance
Real-world Example:
A Clinical Data Coordinator supports clinical trials by reviewing and coordinating study data to ensure it is accurate, consistent, and inspection-ready. This role covers data review, query coordination, safety data alignment, documentation support, and database lock readiness across the trial lifecycle.
What qualifies as the principal diagnosis
Which secondary diagnoses are reportable
Whether conditions were present on admission or developed later
One wrong decision here doesn’t cause a minor denial. It can shift the DRG entirely and trigger audits.
2.Outpatient Medical Coding (OP)
Outpatient medical coding covers patient encounters where there is no overnight admission. These are high-volume, encounter-based services such as clinic visits, OPDs, and same-day procedures.
Here, speed matters, but accuracy matters more because outpatient claims are aggressively reviewed by payers.
This role uses ICD 10 CM coding, CPT coding, and HCPCS coding extensively.
Core Responsibilities
Code individual visits rather than full hospital stays
Assign diagnosis codes that justify medical necessity
Code procedures and services accurately
Apply modifiers correctly
Ensure diagnosis-to-procedure linkage
Follow payer-specific outpatient coding rules
Real-world Example:
A patient visits a clinic for diabetes follow-up and receives lab tests and medication management.
The outpatient coder must ensure:
The visit level matches documentation
Diagnosis codes support the services billed
Procedures are correctly linked
A missing linkage or incorrect modifier doesn’t look dramatic, but it leads to silent denials and rework.
3.Emergency Department Coding (ED)
Emergency department coding deals with urgent and unplanned care. Documentation is often brief, incomplete, and created while treatment is still happening.
Despite short encounters, ED coding carries high audit risk because visit-level coding is heavily scrutinized.
This role relies on ICD 10 CM coding, CPT coding, and HCPCS coding.
Core Responsibilities
Review triage notes, physician documentation, and treatment records
Assign appropriate visit-level codes
Code emergency procedures accurately
Reflect the severity of the patient’s condition
Work within tight turnaround times
Maintain compliance despite limited documentation
Real-world Example
A patient arrives with chest pain, undergoes rapid evaluation, tests rule out a heart attack, and is discharged.
The ED coder must decide:
What level of evaluation and management applies
Whether documentation supports the billed severity
Which diagnoses are reportable
Overcoding attracts audits. Undercoding loses revenue. There’s no comfort zone.
4 . Ancillary Medical Coding
Ancillary medical coding focuses on diagnostic and technical services, not direct patient visits. This includes labs, radiology, pathology, anesthesia, and similar departments.
This role is precision-driven and largely technical, relying heavily on CPT coding and HCPCS coding.
Core Responsibilities
Review test orders and diagnostic reports
Code technical services accurately
Apply modifiers where required
Ensure services match physician orders
Capture all billable services correctly
Real-world Example:
A patient undergoes multiple lab tests and imaging services on the same day.
The ancillary coder must ensure:
Every test performed is captured
Modifiers reflect how services were delivered
Nothing billable is missed
Errors here don’t cause denials immediately. They cause revenue leakage, which is worse because it often goes unnoticed.
Why This Role Separation Matters
Across all four roles, medical coding job responsibilities directly affect:
Claim outcomes
Audit exposure
Compliance standing
Healthcare revenue cycle performance
This is why medical coding accuracy and medical coding compliance are foundational expectations, not optional skills. Each role tests these skills differently.
AI-augmented Medical Coding and
Revenue Intelligence Certification
Master the intersection of traditional medical coding and cutting-edge AI integration. This isn’t just about memorizing books; it’s about leveraging AI tools to automate workflows, enhance precision, and speed up the coding process in real-world clinical environments. Learn to manage complex data sets while maintaining the high compliance standards required by modern global healthcare systems.
AI-Driven Coding Tools, Automated Medical Indexing, ICD-10-CM/PCS & CPT, Natural Language Processing (NLP) in Healthcare, Data Privacy & Ethics, AI-Assisted Auditing, Electronic Health Record (EHR) Optimization
A medical coder’s day does not follow a single routine. What changes the day completely is which type of coding role the coder works in. While the core responsibility remains accuracy and compliance, the pace, decision pressure, and documentation depth vary sharply across inpatient, outpatient, emergency department, and ancillary medical coding.
This section shows how the roles and responsibilities discussed above translate into actual day-to-day work.
1.Starting the Day: Work Queues Look Different by Role
A medical coder does not “open random charts.” They open role-specific work queues.
Inpatient coders start their day with discharge charts from recent hospital stays. These are fewer in number but heavy in documentation.
Outpatient coders see long queues of clinic visits, OPD encounters, and same-day procedures.
Emergency department coders receive time-sensitive emergency encounters that must be coded quickly.
Ancillary coders work through structured queues of lab tests, imaging studies, pathology cases, or anesthesia records.
Right from the first hour, the difference in medical coding job responsibilities is visible.
2.Reviewing Clinical Documentation: Depth vs Speed
The biggest chunk of a coder’s day is documentation review, but how deep that review goes depends on the role.
In inpatient medical coding, coders read admission notes, progress notes, operative reports, and discharge summaries to understand how the patient’s condition evolved.
In outpatient medical coding, coders review encounter notes and procedure documentation focused on that single visit.
In emergency department coding, coders work with brief, evolving documentation created during urgent care.
In ancillary medical coding, coders review test orders, technical reports, and diagnostic results rather than physician narratives.
This is where clinical understanding matters. The responsibility is not to read everything, but to read what affects coding decisions.
3.Identifying Gaps and Risks in Documentation
Real-world documentation is rarely perfect. A key part of daily work is spotting issues before codes are assigned.
Inpatient coders look for missing specificity, unclear diagnoses, or sequencing issues.
Outpatient and ED coders check whether documentation supports the level of service billed.
Ancillary coders verify that services performed match physician orders and reports.
This step protects medical coding compliance. Guessing is not allowed. If documentation does not support the service, the coder flags it.
4.Assigning Codes: Where Accuracy Is Tested
Once documentation is reviewed, the coder applies the appropriate codes.
ICD 10 CM coding is used across all roles for diagnoses.
ICD-10-PCS is applied by inpatient coders for hospital procedures.
CPT coding and HCPCS coding dominate outpatient, emergency, and ancillary work.
This is where medical coding accuracy becomes non-negotiable. Every incorrect code can lead to denials, audits, or revenue loss. Coders must balance correctness with productivity expectations specific to their role.
5.Balancing Accuracy, Compliance, and Productivity
Every coder works under pressure, but the pressure feels different in each role.
Inpatient coders handle fewer charts but face high financial and audit impact.
Outpatient coders manage high volumes with strict payer scrutiny.
Emergency department coders work against turnaround time with limited documentation.
Ancillary coders focus on precision to avoid missed charges and revenue leakage.
Across all roles, the coder’s decisions directly affect the healthcare revenue cycle. This is why medical coding is not data entry; it is a judgment-driven role.
Conclusion
Medical coding is not a single, uniform job. Across inpatient, outpatient, emergency department, and ancillary settings, medical coder roles and responsibilities differ in documentation depth, decision pressure, work pace, and compliance risk. What remains constant across all roles is the requirement for strong clinical understanding, high medical coding accuracy, and strict medical coding compliance. Coding decisions directly influence claim outcomes, audits, and the healthcare revenue cycle.
As healthcare systems evolve, medical coders are expected to move beyond basic code assignment and develop a deeper understanding of workflows, documentation risks, and revenue impact. This shift is further accelerated by the use of automation and AI-driven tools across healthcare revenue cycle operations.
To support learners at different stages, CliniLaunch Research Institute offers two structured learning paths. The Advanced Diploma in Medical Coding focuses on building strong fundamentals in coding guidelines, clinical documentation analysis, and role-based coding practices across inpatient, outpatient, emergency, and ancillary settings. For learners looking to work at the intersection of coding, data, and automation, the AI-augmented Medical Coding and Revenue Intelligence Course goes a step further by introducing AI-assisted workflows, compliance intelligence, and revenue-focused decision-making.
Understanding how medical coding roles differ helps learners choose the right path, set realistic expectations, and build skills that remain relevant in modern healthcare environments.
FAQs
1. Is medical coding just a data entry job?
No. Medical coding requires interpreting clinical documentation, applying guidelines, and using judgment to ensure accuracy, compliance, and correct billing.
2. What is the difference between IP, OP, ED, and Ancillary medical coding?
The difference depends on where care is delivered. Inpatient covers hospital stays, outpatient handles clinic visits, ED focuses on urgent care, and ancillary coding supports diagnostic services.
3. Which medical coding role is best for beginners?
Outpatient coding is the most common starting point for beginners due to structured documentation and encounter-based workflows.
4. Do all medical coders use the same coding systems?
No. All coders use ICD-10-CM, but inpatient coders also use ICD-10-PCS, while outpatient, ED, and ancillary coders mainly use CPT and HCPCS.
5. Can medical coders move into advanced roles later?
Yes. With experience, coders can move into inpatient coding, QA, HCC, or CDI roles.
6. Why is it important to understand medical coding roles early?
Early role awareness reduces confusion during learning, helps choose the right path, and builds realistic career expectations.
A Clinical Research Coordinator (CRC) supports the execution of clinical trials at the study site. They help coordinate patient visits, manage study documents, support informed consent, assist with data collection, and ensure the study follows the approved protocol and regulatory guidelines.
Imagine a hub where scientific protocols, patient care, documentation, sponsor expectations, and compliance all intersect—that’s where a Clinical Research Coordinator (CRC) comes in. CRCs are the operational heart of clinical studies, ensuring that trials are conducted ethically, efficiently, and in strict accordance with regulations and protocols. Clinical trials are the backbone of modern medicine, and behind every successful trial is a skilled professional managing clinical research coordinator roles and responsibilities to keep studies on track.
Research on clinical trial workforce trends shows that the number of registered clinical trials has increased by over 30%, leading to a growing demand for skilled coordinators. This rise highlights the critical role CRCs play in managing trial complexity and supporting timely, high-quality research outcomes.
In this blog, you’ll discover Clinical Research Coordinator Roles and Responsibilities, what they do every day, and how they manage critical aspects like patient visits and study coordination. Whether you’re a student considering a career in clinical research, a team lead wanting to understand your CRC better, or a manager seeking to optimize your study operations, this guide offers practical clarity on clinical research coordinator roles and responsibilities.
Who is clinical Research coordinator?
A Clinical Research Coordinator (CRC) supports the daily conduct of a clinical trial at the study site. They coordinate study activities, manage documentation, and ensure procedures are followed according to the approved protocol. While the principal investigator oversees medical decisions, the CRC handles site-level coordination, so the trial runs smoothly and in compliance.
CRCs work closely with investigators, study staff, sponsors, and the ethics committee to keep communication clear and timely. They help track study timelines, support patient screening and recruitment, and maintain records, so data remains accurate and audit-ready an essential part of CRC responsibilities in clinical trials.
By managing documentation flow, regulatory requirements, patient coordination, and day-to-day trial activities, the clinical research coordinator roles and responsibilities are central to consistent trial conduct and data accuracy. In many research settings, especially within CRC role in hospitals, CRCs support safety reporting compliance and essential documents maintenance, strengthening overall site operations. Evidence from clinical research settings shows that over 80% of sites report improved trial quality and execution when supported by a dedicated CRC, highlighting the importance of effective job role execution and strict ICH-GCP compliance at the site level. This highlights the importance of the clinical research coordinator job role in maintaining operational control and regulatory alignment at the study site.
Roles and Responsibilities of a Clinical Research Coordinator
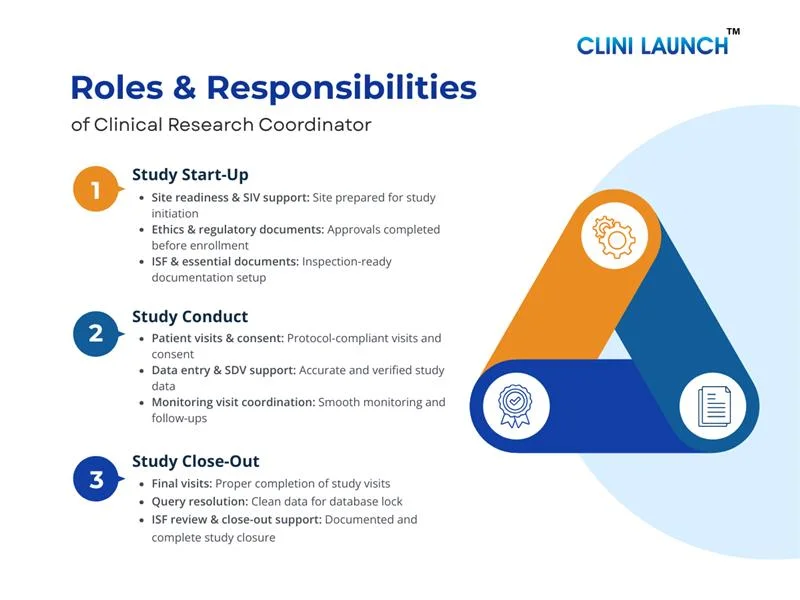
This section explains the clinical research coordinator roles and responsibilities across study start-up, conduct, and close-out phases, reflecting real-world expectations outlined in a standard clinical research coordinator for job description.
Phase 1: Study Start-Up Phase
The study start-up phase prepares the site before enrollment begins. During this stage, the CRC organizes site readiness and ensures alignment with protocol and compliance requirements.
Site Readiness and Study Initiation Support
The CRC supports site readiness by coordinating internal activities, organizing study materials, and assisting with preparation for the Site Initiation Visit (SIV). This ensures the site understands study workflows and is operationally prepared before the trial begins.
Regulatory and Ethics Documentation Support
During start-up, the CRC assists with regulatory and ethics documentation by compiling required records, tracking submission status, and maintaining approval-related correspondence. This helps ensure that all necessary approvals are in place before participant enrollment.
Essential Study Documentation Setup
CRCs organize the trial master file, ISF, delegation logs, training records, and approvals—ensuring inspection of readiness from day one. This ensures documentation is complete, current, and inspection-ready from the start of the study.
CRC Responsibilities During the Study Start-Up Phase
CRC Activity
Purpose
What It Ensures
Site readiness and study initiation support
Prepare the site before trial initiation
Operational readiness and clear workflows
Regulatory and ethics documentation support
Complete required submissions and approvals
Compliance before participant enrollment
Essential study documentation setup
Organize ISF and key study records
Inspection-ready documentation
Case Study: Impact of Protocol Deviations on Clinical Trial Quality
What Happened
Impact
Why CRC Matters
Clinical research literature shows that protocol deviations—departures from the approved study procedures—
frequently occur during the active conduct phase of clinical trials and can compromise both participant
safety and the reliability of trial data. These deviations may include missed assessments, improper
documentation, or procedures conducted outside the defined protocol.
When protocol deviations are not properly identified, recorded, and managed, they can undermine the
scientific validity of a trial, affect data integrity, and potentially jeopardize patient welfare. This
can lead to increased monitoring findings, longer trial completion times, and difficulties during
regulatory review if deviations are widespread or poorly documented.
During the study conduct phase, Clinical Research Coordinators (CRCs) help minimize protocol deviations by
ensuring daily trial activities strictly follow the approved protocol, supporting accurate record-keeping,
and coordinating with monitors and investigators to address discrepancies quickly. Their ongoing oversight
is key to maintaining data quality and overall study integrity.
Phase 2: Study Conduct Phase
CRCs support active trial execution and ensure ongoing compliance throughout the conduct phase. CRCs also support the clinical research coordinator for job description by assisting with adverse event reporting and serious adverse event reporting in coordination with the investigator and sponsor.
Patient Visit Coordination and Study Activities
The CRC coordinates participant visits according to the study schedule by arranging appointments, preparing visit-related materials, and supporting site staff during study procedures. This helps ensure visits are conducted on time and in line with protocol requirements.
Informed Consent and Participant Support
CRCs support the informed consent process and informed consent documentation, ensuring ethical participation and compliance. The CRC also assists with participant communication to support adherence to visit schedules and study requirements.
Data Collection and Documentation Maintenance
CRCs maintain source documentation, complete case report forms, and support data entry and data accuracy. This helps maintain data quality and consistency throughout the study.
Monitoring Visit and Sponsor Coordination
CRCs support monitoring visit support, SDV activities, and maintain audit and inspection of readiness, ensuring study documents are available for review, and addressing follow-up actions. This helps maintain ongoing oversight and inspection of readiness during the study.
CRC Responsibilities During the Study conduct Phase
CRC Activity
Purpose
What It Ensures
Patient visit coordination and study activities
Manage scheduled study visits and protocol-required procedures
Protocol-compliant and timely study visits
Informed consent and participant support
Maintain valid, informed, and ongoing consent
Ethical participant involvement and regulatory compliance
Data collection and documentation maintenance
Record, verify, and manage study data accurately
Data quality, traceability, and consistency
Monitoring visit and sponsor coordination
Support sponsor monitoring, audits, and oversight activities
Inspection readiness and timely issue resolution
What Happened
Impact
Why CRC Matters
During the conduct phase of clinical trials, loss to follow-up occurs when enrolled participants fail to
return for scheduled study visits or withdraw prematurely. Clinical research methods literature indicates
that approximately 20% of subjects may be lost to follow-up, which can introduce bias and misleading
results, particularly when missing outcomes differ between treatment groups.
High rates of loss to follow-up can distort treatment effect estimates, threaten the internal validity of
trial results, and necessitate additional statistical adjustments or sensitivity analyses. These issues
complicate data interpretation and may raise concerns during regulatory review and acceptance.
Clinical Research Coordinators (CRCs) play a key role in minimizing loss to follow-up by maintaining
participant engagement, proactively tracking visit schedules, following up with participants, and
documenting reasons for missed visits. Their efforts help protect data completeness, reduce bias, and
preserve the overall quality and validity of trial data.
Phase 3: Study Close-Out Phase
The study close-out phase begins after the last participant completes the final study visit and continues until all study activities at the site are formally completed. During this stage, the Clinical Research Coordinator (CRC) supports the completion of site-level activities by ensuring documentation is finalized, data is resolved, and the site is ready for study closure and potential inspections.
Final Patient Visit and Study Completion Support
The CRC supports final study visits by coordinating end-of-study assessments, ensuring required procedures are completed, and confirming that participant records are properly closed. This helps ensure that all subject-related activities are completed in line with the protocol.
Data Cleaning and Query Resolution
During close-out, the CRC supports data cleaning by responding to outstanding data queries, verifying source documents, and assisting with final Case Report Form (CRF) completion. This helps ensure that data is accurate, complete, and ready for database locking.
Essential Document Review and Archival Preparation
The CRC reviews the Investigator Site File (ISF) to ensure all required documents are complete, current, and properly filed. This includes confirming approvals, correspondence, and study records are ready for long-term storage according to regulatory requirements ensure records meet essential documents of maintenance standards.
Close-Out Visit and Sponsor Coordination
The CRC supports site close-out visits by coordinating with Clinical Research Associates (CRAs), making documents available for review, and addressing close-out findings. This helps ensure that all site responsibilities are formally completed and documented.
CRC Responsibilities During the Study Close-Out Phase
CRC Activity
Purpose
What It Ensures
Final patient visit coordination
Support completion of end-of-study and follow-up visits
Proper subject closure in accordance with the protocol
Data cleaning and query resolution
Address outstanding data issues and resolve queries
Readiness for database lock and analysis
Essential document review (ISF)
Verify completeness and accuracy of study documentation
Inspection-ready study records
Close-out visit support and coordination
Assist the CRA during site close-out activities
Formal completion of site-level study activities
What Happened
Impact
Why CRC Matters
The U.S. FDA issued a warning letter to a clinical investigator for failing to retain required study records
for the mandated retention period. Record retention is a critical responsibility during the study
close-out and archiving phase, and failure to meet these requirements represents a significant regulatory
non-compliance.
Inadequate or missing record retention raises serious concerns regarding the validity, reliability, and
integrity of site-level trial data. Such deficiencies can result in corrective and preventive actions
(CAPAs), heightened regulatory scrutiny, and potential restrictions on future research activities.
During the close-out phase, Clinical Research Coordinators (CRCs) help prevent record retention failures by
reconciling essential documents, ensuring proper archiving, maintaining document traceability, and
confirming that records remain accessible for inspections throughout the required retention period. Their
oversight helps keep the site inspection-ready even after trial completion.
Skills Required for a Clinical Research Coordinator (CRC)
A Clinical Research Coordinator requires a strong foundation in clinical research and technical skills to support compliant trial execution. This includes understanding clinical trial protocols, ICH-GCP compliance, informed consent handling, source data verification, and proper management of essential study documents such as the Investigator Site File (ISF). These skills ensure trials are conducted ethically, data remains accurate, and sites remain inspection-ready throughout the study’s lifecycle.
In addition to technical knowledge, CRCs need operational and professional skills to manage daily trial activities effectively. This includes coordinating study visits, working closely with investigators, CRAs, and study teams, resolving data queries, tracking timelines, and maintaining clear communication. Strong attention to detail, time management, and ethical awareness enable CRCs to handle multiple responsibilities while maintaining compliance and consistent trial performance.
Conclusion
A CRC’s daily work directly affects trial quality, patient safety, and data integrity. From documentation to coordination, clinical research coordinator roles and responsibilities are central to ethical and efficient research execution. Every task, whether managing patient visits, maintaining documentation, or supporting compliance, directly affects the quality, safety, and credibility of a clinical trial. This makes the CRC role essential to ensuring that research studies are conducted responsibly and efficiently at the site level.
For those exploring a career in clinical research, understanding the CRC role provides clarity on what real trial execution looks like beyond theory. To learn more about this role in depth, explore the skills that define the CRC blogon the CliniLaunch website, and for hands-on, job-ready training, consider enrolling in the Advanced Diploma in Clinical Research to build the skills required to succeed in this field.
FAQ
1. What are the roles and responsibilities of a Clinical Research Coordinator?
A Clinical Research Coordinator supports the day-to-day conduct of clinical trials at the site. They coordinate study activities, manage documentation, support patient visits, and ensure the trial follows the approved protocol and GCP requirements.
2. What does a Clinical Research Coordinator do in a clinical trial?
A CRC coordinates patient visits, maintains study records, supports data entry, and works with investigators and monitors to keep the trial running smoothly and compliantly throughout its lifecycle.
3. What is the difference between a CRC and a CRA?
A CRC works at the study site handling daily trial execution, while a CRA works for the sponsor or CRO to monitor sites, review data, and ensure compliance across multiple sites.
4. What is the difference between a CRC and a CRA?
A CRC typically reports to the Principal Investigator (PI) at the site and works closely with Clinical Research Associates (CRAs) for study of coordination and monitoring activities.
5. What qualifications are required to become a Clinical Research Coordinator?
Most CRCs have a background in life sciences, pharmacy, nursing, or healthcare. Formal training in clinical research and GCP knowledge significantly improves eligibility and readiness for the role.
6. What skills are needed for a Clinical Research Coordinator?
CRCs need strong protocol understanding, documentation skills, attention to detail, coordination ability, and clear communication skills to manage study activities and compliance effectively.
7. What is the role of a CRC in patient safety?
CRCs support patient safety by ensuring informed consent is properly documented; adverse events are reported on time, and study procedures are followed as per protocol and ethical guidelines.
8 . Does a Clinical Research Coordinator take informed consent?
A CRC supports the informed consent process by explaining study details and documenting consent, but the investigator retains final responsibility for ensuring consent is ethically and medically appropriate.
9. Is Clinical Research Coordinator a good career option?
Yes, the CRC role offers hands-on exposure to clinical trials, strong demand across hospitals and research sites, and a solid foundation for long-term growth in clinical research.
10 . What is the career growth path for a Clinical Research Coordinator?
With experience, CRCs can progress to roles such as Senior CRC, CRA, Clinical Trial Manager, or Project Coordinator, depending on skills, exposure, and career interests.
A Clinical Data Coordinator supports clinical trials by reviewing and coordinating study data to ensure it is accurate, consistent, and inspection-ready.
This role covers data review, query coordination, safety data alignment, documentation support, and database lock readiness across the trial lifecycle.
The Clinical Data Coordinator is one of the most critical entry-level roles in clinical research for anyone aiming to build a non-laboratory career in healthcare. As clinical trials become increasingly data-driven, multi-site, and tightly regulated, this role exists to protect one thing the industry cannot afford to lose: data integrity.
Every clinical trial generates massive volumes of patient data. That data must be accurate, consistent, traceable, and compliant with global regulatory standards. If it is not, the trial risks delays, audit findings, inspection observations, or outright failure. The clinical data coordinator plays a central role in preventing those outcomes by ensuring trial data is review-ready, compliant, and reliable throughout the study lifecycle.
For students exploring a career in clinical research, understanding this role is often the first practical step toward entering the industry without working at the lab bench.
Who is a Clinical Data Coordinator?
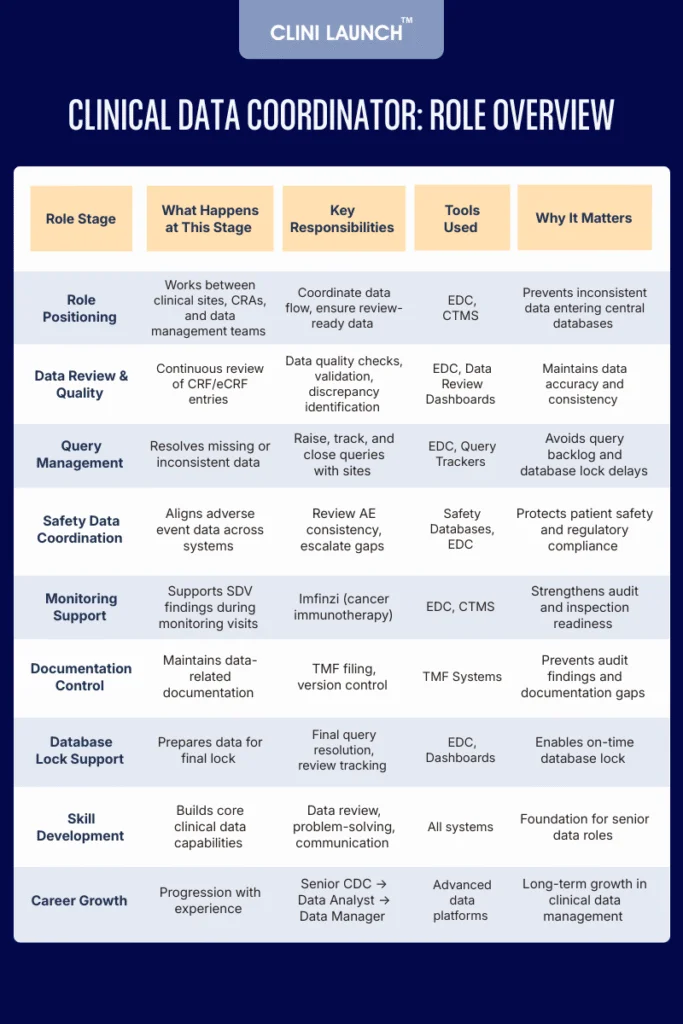
A Clinical Data Coordinator (CDC) is a clinical research professional who supports clinical data management activities during a clinical trial. This role does not generate patient data. Instead, it focuses on reviewing, validating, and coordinating data collected from clinical sites to ensure it meets protocol, quality, and regulatory requirements.
CDC works closely with clinical research associates, investigators, site teams, and data managers. Their job is to make sure data entered into electronic data capture systems is accurate, complete, consistent, and aligned with the study protocol.
In practical terms, this role sits between data collection at the site level and centralized data management. By coordinating data flow across teams and systems, the clinical data coordinator ensures that issues are identified early, queries are handled properly, and patient safety data remains consistent across the trial.
For beginners, this role offers structured exposure to how real clinical trials operate from a data quality, compliance, and regulatory perspective, making it a common and logical entry point into clinical research careers.
Core Responsibilities of a Clinical Data Coordinator
The core responsibilities of a Clinical Data Coordinator focus on ensuring that clinical trial data is accurate, complete, traceable, and compliant throughout the study lifecycle. This role supports the smooth flow of data from clinical sites to central databases while maintaining regulatory alignment and database readiness.
Below is a clear and beginner-friendly explanation of clinical data coordinator roles and responsibilities, supported by real-world, industry-based examples.
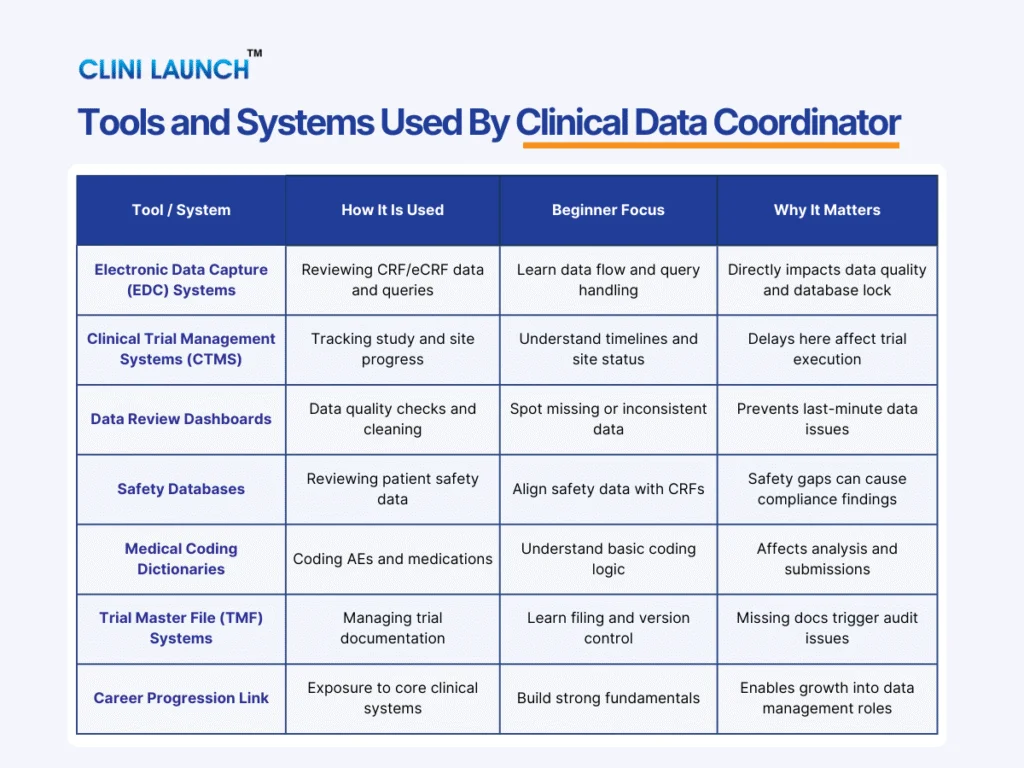
1.Supporting Clinical Data Management Activities
Clinical trials follow a predefined data management plan that defines how data should be collected, reviewed, cleaned, and locked. The Clinical Data Coordinator supports this plan by ensuring that daily data workflows are executed consistently across the study. This includes coordinating data review cycles, tracking data-related issues, and working closely with central data management teams to resolve inconsistencies early rather than at the end of the trial.
2.Case Report Forms and eCRF Handling
Case report forms are the primary tools used to capture patient data in a clinical trial. In most modern studies, this data is entered through electronic case report forms using electronic data capture systems. The Clinical Data Coordinator ensures that these forms are completed accurately and in line with the approved protocol. This involves reviewing form completeness, verifying required fields, coordinating updates after protocol amendments, and supporting corrections when inconsistencies are identified. Proper CRF and eCRF handling are essential for maintaining data consistency and accuracy across study sites.
Advanced Diploma in
Clinical Research
Build practical, industry-aligned skills to work across real clinical trial environments. Learn how clinical studies are planned, conducted, documented, and monitored, with a strong emphasis on ethics, patient safety, and regulatory compliance throughout the trial lifecycle.
Although patient data is entered by clinical sites, the Clinical Data Coordinator plays a key role in reviewing and validating that data before it progresses through the data lifecycle. This includes checking entries in electronic data capture systems for missing, illogical, or inconsistent values and supporting structured data entry and validation of workflows. When recurring issues are identified, they are escalated to data managers to prevent systemic data quality problems. These activities contribute directly to early-stage data cleaning in clinical trials.
4.Query Management and Data Cleaning
When discrepancies or missing information are identified during data review, data queries are raised for clarification. The Clinical Data Coordinator supports query management by reviewing system-generated and manual queries, coordinating responses with site teams, and tracking query resolution status. Active involvement in ongoing data cleaning helps prevent query backlogs and reduces delays during database locks.
Real-Life Case Study: How Query Backlogs Delay Database Lock
In a multi-site Phase III clinical trial, data queries were raised regularly for missing and inconsistent entries, but follow-ups with sites were delayed. As the study approached database lock, unresolved queries accumulated, slowing final data cleaning and putting the lock timeline at risk.
The Clinical Data Coordinator intervened by prioritizing high-impact queries, coordinating closely with sites and Clinical Research Associates, and enforcing structured tracking with clear timelines. This focused approach cleared the backlog, enabled database lock on schedule, and prevented delays to final analysis and regulatory activities.
Regulatory guidance from the U.S. FDA emphasizes timely data review, correction of discrepancies, and readiness of electronic source data before database lock and submission.
5.Source Data Verification Support
During monitoring visits, Clinical Research Associates perform source data verification by comparing site source documents with data entered the clinical database. The Clinical Data Coordinator supports this process by clarifying discrepancies raised during monitoring, coordinating corrections with sites, and ensuring related documentation is updated correctly. This support strengthens audit and inspection of readiness and improves overall trial quality.
6. Patient Safety Data Coordination
Patient safety data must be accurate, timely, and consistent across systems to meet regulatory expectations. The Clinical Data Coordinator supports patient safety data coordination by verifying adverse event entries, ensuring consistency between safety databases and case report forms, escalating missing or delayed safety data, and aligning safety information with regulatory reporting timelines.
Real-Life Case Study: Inconsistent Patient Safety Data Across Systems
During routine data review in an ongoing clinical trial, inconsistencies were identified between adverse events recorded in case report forms and entries in the safety database. If left unresolved, these discrepancies could have resulted in compliance findings during regulatory inspection.
The Clinical Data Coordinator reviewed safety data entries, coordinated corrections with study sites, and ensured alignment between clinical and safety systems. As a result, patient safety data became consistent across platforms; regulatory reporting timelines were met, and inspection risk was reduced.
Regulatory inspection trend reports published by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) frequently cite safety data inconsistencies and documentation gaps as inspection observations.
7. Regulatory Compliance and ICH GCP Alignment
All clinical trial data activities must align with ICH GCP compliance and applicable regulatory requirements. The Clinical Data Coordinator ensures protocol adherence during data review, proper documentation for audits, and compliance with regulatory expectations. The coordinator also supports sponsor reviews and inspections by maintaining inspection-ready data and documentation throughout the trial.
8.Trial Master File and Documentation Support
Proper documentation is essential for trial transparency and regulatory inspections. The Clinical Data Coordinator supports trial master file activities by ensuring that data-related documents are filed correctly, version control is maintained, and documentation practices remain inspection-ready throughout the study.
Real-Life Case Study: Preventing Audit Findings Through Proper Documentation
During a sponsor audit, missing data review documentation was identified for several completed study visits. Although the clinical data itself was accurate, incomplete documentation raised concerns about data traceability and inspection readiness.
The Clinical Data Coordinator reviewed filing practices, ensured missing documents were added to the trial master file, and standardized documentation workflows. The audit observations were resolved successfully, and improved documentation practices were applied to ongoing and future studies.
Regulatory inspection summaries published by the U.S. Food and Drug Administration (FDA) consistently highlight documentation gaps and incomplete records as common audit findings in clinical trials.
9.Database Lock Support
Before a clinical trial database can be locked, all data queries must be resolved, and final data reviews completed. The Clinical Data Coordinator supports database lock activities by tracking outstanding queries, confirming site responses, supporting final review cycles, and coordinating with data managers prior to lock approval. This ensures that the database is complete, accurate, and ready for submission.
10.Medical Coding Coordination
Some clinical trials require standardized coding of medical terms such as adverse events and medications. The Clinical Data Coordinator assists in medical coding coordination by supporting review of coded terms, coordinating corrections when inconsistencies are identified, and ensuring consistency across datasets used for analysis and reporting.
11. Collaboration with Clinical Teams
Clinical trials depend on effective coordination across multiple teams. The Clinical Data Coordinator works closely with Clinical Research Associates, investigators, site staff, and data managers to resolve data-related issues, support site communication, and ensure smooth operational flow throughout the trial.
Together, these responsibilities ensure that clinical trial data is reliable, audit-ready, and suitable for regulatory submission, reinforcing the Clinical Data Coordinator’s role as a critical link between clinical sites and data management teams.
Key Skills and Career Growth for a Clinical Data Coordinator
Key Skill Area
What It Enables in the Role
How This Skill Is Built
Career Growth It Supports
Attention to Detail & Data Review
Identifying missing, inconsistent, or incorrect data
Regular data review, query checks, and CRF verification
Progression to Senior Clinical Data Coordinator
Clinical Data Management Knowledge
Working with CRFs, eCRFs, and data review cycles
Exposure to data management plans and review workflows
Transition into Clinical Data Analyst or Data Manager
Regulatory & ICH GCP Awareness
Ensuring compliant data handling and audit readiness
Working with protocols, GCP guidelines, and inspections
Eligibility for lead and compliance-focused roles
Query Management & Issue Resolution
Coordinating with sites and CRAs to close data queries
Handling live queries and site clarifications
Study-level ownership and senior coordinator roles
Problem-Solving Ability
Identifying data issues and driving corrective actions
Managing discrepancies and recurring data issues
Readiness for complex trials and lead roles
Communication & Coordination
Working with investigators, CRAs, and data teams
Daily interaction with cross-functional teams
Growth into Clinical Data Operations roles
Documentation & Process Tracking
Maintaining inspection-ready records
Managing trackers, TMF documents, and audit files
Supports management and oversight responsibilities
Advanced Diploma in
Clinical SAS
Build practical skills in clinical data analysis and statistical reporting using SAS, aligned with regulatory standards used in clinical trials. Learn how clinical trial data is structured, analyzed, and converted into submission-ready outputs.
The Clinical Data Coordinator role exists to keep a clinical trial under control. Modern trials involve multiple sites, large volumes of patient data, and strict regulatory oversight. When data is not reviewed, coordinated, and resolved in real time, problems surface late, during audits, inspections, or database lock. This role prevents that by acting as the link between sites, clinical teams, and data management.
For beginners, the value of this role is exposure. You see how trials actually function, how data flows, where mistakes happen, and how those mistakes are corrected before they become regulatory issues. You are not isolated in one task; you are embedded in the operational backbone of a trial.
To enter and succeed in this role, theoretical knowledge alone is not enough. What matters is practical familiarity with clinical trial workflows and regulatory expectations. Structured, industry-aligned training helps bridge that gap and prepares candidates to operate confidently in real clinical research environments.
Clini Launch Research Institute offers an industry-aligned clinical research course that equips learners with the skills and hands-on exposure required to confidently begin a career as a Clinical Data Coordinator and grow within the clinical research industry.
FAQs
1. What does a Clinical Data Coordinator do?
A Clinical Data Coordinator reviews and coordinates clinical trial data to ensure it is accurate, complete, and compliant with study protocols and regulatory guidelines.
2. Is the Clinical Data Coordinator role suitable for beginners?
Yes. It is an entry-level role in clinical research and is suitable for life science graduates who want a non-laboratory career path.
3. Does a Clinical Data Coordinator perform data entry?
No. Data is entered by clinical sites. The coordinator reviews, validates, and resolves data issues.
4. What tools does a Clinical Data Coordinator work with?
They commonly work with electronic data capture systems, clinical trial management systems, safety databases, and trial master file systems.
5. What is the career growth after becoming a Clinical Data Coordinator?
With experience, professionals can progress into Senior Clinical Data Coordinator, Clinical Data Manager, or Clinical Data Operations roles.
6. How can I prepare for a Clinical Data Coordinator role?
Preparation involves understanding clinical research processes, basic data management concepts, regulatory guidelines, and gaining practical exposure through structured training.
Informed Consent in Clinical Trials
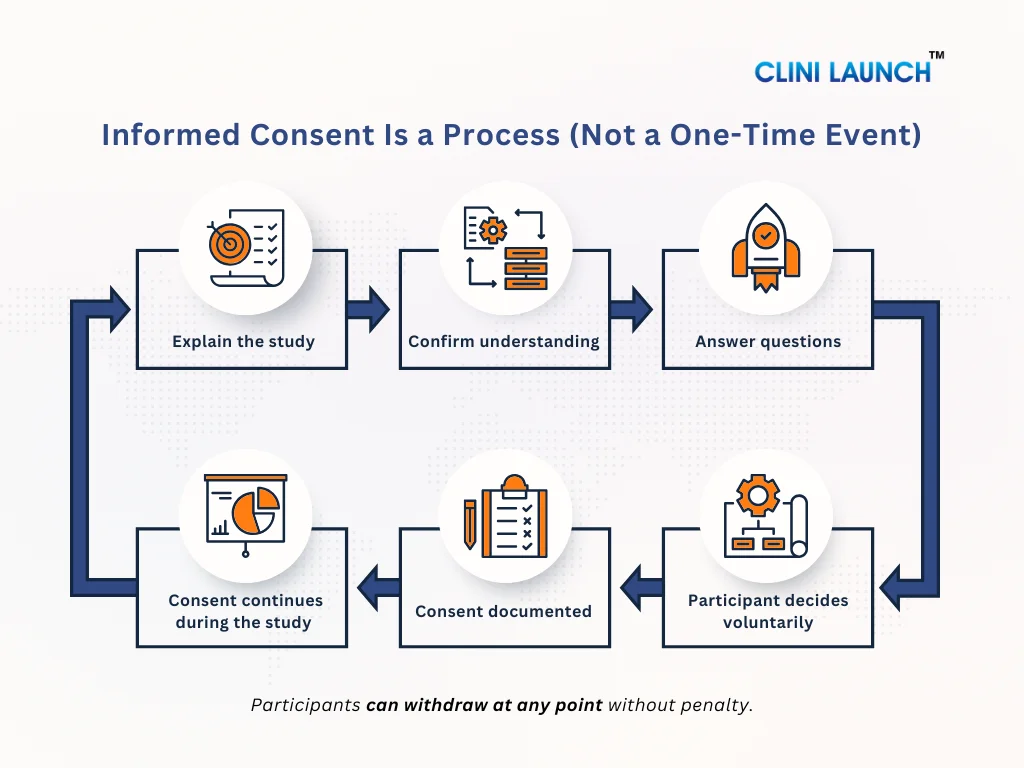
Informed consent in clinical trials is an ongoing process where participants are clearly informed about the study purpose, procedures, potential risks, expected benefits, and their right to withdraw from the study at any time without penalty.
Every clinical trial begins with a single non-negotiable requirement: informed consent. Before a participant can be screened, tested, or exposed to any study-related procedure, their voluntary and informed agreement must be obtained and documented. This is not an administrative formality. It is the ethical and legal foundation of clinical research.
Without valid informed consent, a clinical trial cannot proceed, no matter how well designed the protocol is or how promising the investigational treatment may be. Failures in the consent process have repeatedly led to trial suspensions, regulatory action, and loss of public trust. This is why informed consent is treated as a core ethical responsibility in clinical research, not just a signed document.
What Is Informed Consent in Clinical Trials
In clinical trials, the informed consent process means that a participant clearly understands what a study involves and agrees to take part voluntarily. Before joining a trial, the participant is informed about the study’s purpose, what procedures will take place, the possible risks and benefits, and their right to refuse or withdraw at any time. Only after this information is clearly understood can a participant make an informed decision to participate.
In simple terms, this explains the informed consent process and how it is applied in real clinical trial settings as an ongoing communication process rather than just a signed form.
Informed Consent Is a Process, not a Signature
Many people think informed consent is just signing a form. This is not true. Informed consent is an ongoing process. It starts when the study is first explained and continues throughout the trial. Participants should always be kept informed. If anything changes in the study or new risks are found, the participant must be told again and given a chance to decide whether they still want to continue.
The 3 Core Requirements of Informed Consent
For informed consent to be valid in a clinical trial, it must meet three essential requirements. If even one of these is missing, the consent is considered incomplete. These requirements are central to the informed consent definition used in ethical clinical research.
1. Voluntary
Consent must be given freely and without pressure. The participant should never feel forced, rushed, or afraid to say no.
This means:
Participation is optional
Saying no will not affect medical care
The participant can leave the study at any time
For example, a participant should not feel that joining the trial is the only way to receive treatment or medical attention. True consent exists only when the decision is made by choice.
2. Understandable
The information shared must be easy to understand. Using complex medical terms or speaking too fast can prevent real understanding.
This means:
Information should be explained in simple language
Medical jargon should be avoided or clearly explained
The participant should be encouraged to ask questions
Understanding is not assumed just because someone signs a form. Researchers must make sure the participants truly understand what the study involves.
3. Informed
The participants must receive complete and honest information about the study.
This includes:
Why the study is being done
What will happen during the study
Possible risks and side effects
Possible benefits (or lack of direct benefit)
Other treatment options available
Consent is considered informed only when the participant has enough information to make a thoughtful and confident decision.
Advanced Diploma in
Clinical Research
Build industry-ready skills to work across real clinical trial environments, from study initiation to close-out. Learn how clinical research actually operates in hospitals, CROs, pharma companies, and research organizations, with a strong focus on compliance, documentation, and trial execution.
What Participants Must Be Told During Informed Consent
For informed consent to be valid, participants must be clearly told about certain essential information before they agree to join a clinical trial. This ensures they can decide with confidence, clarity, and without pressure.
Purpose, Duration, and What Will Happen
Participants must be told:
Why the study is being done and what question it aims to answer
How long the study will last, including follow-up periods
What exactly will happen to them, such as clinic visits, tests, treatments, or sample collection
This helps participants understand the level of involvement required and decide whether the study fits their personal and medical situation.
Risks, Discomforts, and Possible Benefits
Participants must be informed about:
Possible risks and side effects, even if they are uncommon
Any physical, emotional, or practical discomfort they may experience
Possible benefits, if any, and whether there may be no direct benefit to them
Risks must be explained honestly and clearly so participants can balance potential harm against possible benefits before deciding.
Alternatives to Participation
Participants must be told:
Other available treatment or care options
That choosing not to participate will not affect their access to medical care
This ensures participants do not feel pressured to join the study or believe it is their only option.
Privacy, Confidentiality, and Data Use
Participants must understand:
How their personal and medical information will be collected and stored
Who may access their data, such as researchers or regulatory authorities
How their identity will be protected as far as possible
These build trust and reassure participants that their personal information will be handled responsibly.
Costs, Compensation, and Treatment for Injury
Participants must be told:
Whether there are any costs related to participating in the study
If compensation or reimbursement is provided for time or travel
What medical care or compensation is available if a study-related injury occurs
Clear communication in this area helps prevent confusion or disputes later.
Right to Refuse or Withdraw Without Penalty
Participants must be clearly told that:
Participation in the study is completely voluntary
They can refuse to participate without giving a reason
They can withdraw from the study at any time without losing medical care or benefits
This reinforces participant’s autonomy and ensures they remain in control of their decision.
Whom to Contact for Questions and Emergencies
Participants must be given:
Contact details of the study investigator or study team
Information on whom to contact for general questions
Emergency contact details for urgent situations
This ensures participants know where to seek help or clarification at any stage of the study.
How the Consent Process Works in Real Clinical Trials
In real clinical trials, informed consent is not a one-time formality. It is a structured, step-by-step process designed to protect participants before they join a study and while the study is ongoing. Regulatory authorities such as the U.S. Food and Drug Administration clearly state that informed consent must be obtained before any study-related activity begins and must continue throughout the trial. This section shows informed consent processes in real clinical trials, beyond theory and documentation.
1. When Consent Happens — Before Any Study Procedure
In clinical trials, informed consent must be obtained before:
Any screening tests
Any study-related examination
Any trial medication or intervention
Any data or sample collection
The study team explains the study in detail, answers questions, and ensures the participant understands the information. Only after this discussion does the participant sign the informed consent form. If any study procedure happens before consent, the consent is considered invalid.
When Consent Comes Too Late: India’s 2010 Clinical Trial Crisis
In the early 2010s, serious concerns emerged in India regarding how clinical trials were conducted. Reports indicated that some participants were enrolled without fully understanding the study, and in certain cases, informed consent was obtained only after study-related procedures had already begun.
These concerns reached the Supreme Court of India through a public interest litigation filed by Swasthya Adhikar Manch, an organization advocating for patient rights. The court examined whether trial participants were adequately informed before being exposed to investigational drugs or procedures.
The findings were significant. In 2013, the Supreme Court halted approvals for new clinical trials until stricter safeguards were established. A key issue identified was the failure to obtain valid informed consent prior to any study procedure.
This case reinforced a fundamental principle of clinical research: consent obtained after a procedure is not considered valid informed consent.
The outcome reshaped India’s clinical trial oversight framework and demonstrated how failures in informed consent can lead to major legal and regulatory intervention.
2. “Key Information” Upfront — What Matters Most First
In real consent discussions, participants are first given key information that directly affects their decision, such as:
Why the study is being conducted
What will happen to them
Major risks and possible benefits
The voluntary nature of participation
This information is presented before lengthy technical details, so participants can quickly understand what truly matters and decide whether they want to continue learning about the study.
When Key Information Is Not Told First: The Coventry Chapati Study
Another important lesson in informed consent comes from the United Kingdom. In a nutrition study conducted in Coventry, South Asian women were given chapatis containing a radioactive iron isotope to examine iron absorption. Although consent documentation existed, later reviews identified a serious ethical failure.
Many participants were not clearly informed at the outset that radioactive material was involved. The true nature of the study and its potential risks were not explained in a manner the participants could easily understand. Critical information was either minimized or not communicated clearly at the beginning of the consent process.
This study later became widely cited in bioethics discussions because it demonstrated that informed consent fails when key information is hidden, delayed, or deprioritized—even if a consent form is signed.
The Coventry Chapati Study reinforced a fundamental principle of ethical research: participants must receive the most important information first—study purpose, procedures, and risks—before secondary details or paperwork.
The case continues to be referenced as a clear example of why transparency at the very start of the informed consent process is essential.
3. Ongoing Consent During the Study
Informed consent does not end once the form is signed. During the study:
Participants may ask questions at any time
New risks or safety findings must be shared
Protocol changes may require re-consent
Participants must be free to reconsider and withdraw
This ensures that consent remains informed and voluntary throughout the study, not just at the beginning.
When Consent Is Treated as a One-Time Event: HIV Trials in Uganda
A different type of informed consent failure was observed in HIV clinical trials conducted in Uganda. In these studies, participants signed informed consent forms correctly at the time of enrollment, and on paper, the consent process appeared compliant with regulatory requirements.
However, follow-up assessments of participant understanding revealed significant gaps over time. Many participants did not fully understand the study as it progressed. Some were unclear about their right to withdraw, while others misunderstood ongoing study procedures and expectations.
Contributing factors included language barriers, low literacy levels, and long study durations. Without repeated explanations and reinforcement, participant understanding gradually declined. Researchers concluded that a single consent discussion at the beginning of the trial was insufficient.
This led to an important principle in modern clinical research: informed consent is an ongoing process. Participants must be given continuous opportunities to ask questions, receive updates, and reconfirm their willingness to participate—especially when new information or changes arise during the study.
Documentation and Formats in the Informed Consent Process
In clinical trials, informed consent must be documented properly to show that a participant understood the study and agreed to take part before any study procedure began. Proper documentation supports the informed consent form and compliance with ethics committee (IEC/IRB) requirements.
1.Written Consent and Signatures
The study is explained to the participant
The informed consent form is signed and dated
The investigator also signs the form
This signed document is the official proof of consent. It confirms that consent was taken correctly and on time.
2.Witness or Translator (When needed)
Used when a participant cannot read or understand the consent form
A translator explains the study in the participant’s language
A witness confirms the explanation and signs
This ensures that consent is truly understood. It protects participants from agreeing without understanding.
3.Electronic Informed Consent (eConsent)
Consent is taken digitally using a tablet or computer
May include videos or simple explanations
Digital signatures and timestamps are recorded
This helps reduce paperwork errors and improves understanding. It makes the consent process clearer and more reliable.
Advanced Diploma in
Medical Coding
Develop skills in clinical documentation, medical records analysis, and compliance practices that support ethical patient care and informed consent processes across healthcare and research settings. This program focuses on accuracy, standards, and real-world documentation workflows.
Special Situations Beginners Must Know in Informed Consent
In real clinical trials, informed consent does not always happen in a simple situation where an adult participant reads a form and signs it. Certain special situations require extra care to protect participants and ensure that consent remains ethical, valid, and fair.
1.Legally Authorized Representative (When a Participant Can’t Consent)
Sometimes, a participant is unable to give informed consent on their own, such as:
When the participant is unconscious
When there is severe cognitive impairment
When the participant is critically ill
In these situations, consent is obtained from a Legally Authorized Representative (LAR) usually a close family member or legal guardian, as permitted by law. Guidance from the U.S. Department of Health and Human Services makes it clear that this option is used only when the participant truly cannot decide for themselves.
The LAR is expected to act in the best interest of the participant, not for convenience or speed.
2.Assent for Minors
Children cannot legally provide informed consent on their own. When clinical research involves minors:
Consent must be obtained from a parent or legal guardian
The child’s assent (agreement) should also be sought, when appropriate
Assent means explaining the study to the child in age-appropriate, simple language and respecting their willingness or refusal to participate. Ethical guidance from the U.S. Department of Health and Human Services emphasizes that children should be involved in the decision as much as they are able to understand.
3.Vulnerable Participants and Undue Influence
Some participants may be considered vulnerable, such as:
Economically disadvantaged individuals
Patients’ dependent on doctors, caregivers, or institutions
Individuals with limited education or low health literacy
In these cases, extra safeguards are required to ensure participation is truly voluntary. Guidance from the U.S. Food and Drug Administration stresses that consent must not be influenced by fear, authority, financial pressure, or promises of better care.
Participants must clearly understand that saying “no” will not affect their treatment or benefits.
4.Waiver, Alteration, and Emergency Exceptions (High-Level)
In rare situations, informed consent requirements may be waived or altered, such as:
Certain minimal-risk research
Emergency situations where immediate medical action is required
These exceptions are strictly controlled and allowed only with ethics committee approval and under regulations outlined in the eCFR. Consent is not ignored; it is adjusted only when participant’s safety or urgent public health needs require it.
5.Re-Consent After Protocol Changes or New Risk Information
Informed consent is an ongoing process, not a one-time event. If:
The study protocol changes
New risks are identified
New information affects participant safety
Participants must be informed again, and re-consent may be required. Guidance from the ICH makes it clear that participants have the right to reconsider their participation when new information emerges.
Key Takeaway for Beginners
Informed consent is about protecting people, not paperwork.
Even in special or complex situations, consent is never skipped. Instead, it is adapted to ensure fairness, understanding, and respect. This may involve consent from a legally authorized representative, support through a child’s assent, additional safeguards for vulnerable participants, adjustments in emergency settings, or repeating consent after study changes.
Regardless of the situation, the objective remains the same: participants must always have a meaningful choice about whether to take part in the research.
Common Consent Mistakes (And Why They’re Serious)
In clinical research, informed consent usually fails not because of bad intent, but because of small, routine mistakes. For beginners, it’s important to recognize these mistakes early, because even simple errors can make consent ethically invalid and create serious regulatory issues. Many of these mistakes arise from misunderstanding the difference between consent and informed consent.
Using “Too Technical” Language and Poor Understanding
One of the most common consent mistakes is explaining the study using complex medical or scientific language that participants cannot easily understand.
This typically happens when:
Consent forms are written like scientific or regulatory documents
Medical terms are not explained in simple language
The study is explained quickly without checking understanding
Regulatory expectations under the eCFR require that consent information be understandable to the participant. If a participant does not truly understand what they are agreeing to, the consent is not considered informed even if the form has been signed.
Coercion, Pressure, or Misleading Promises
Another serious mistake is influencing participants through pressure, authority, or misleading information, rather than allowing them to decide freely.
This can occur when:
Doctors or study staff unintentionally pressure participants
Participants fear losing medical care if they refuse
Benefits are exaggerated or described as guaranteed
Guidance from the U.S. Food and Drug Administration clearly states that informed consent must be voluntary and free from coercion or undue influence. If a participant agrees because they feel pressured or misled, the consent is no longer voluntary and therefore not valid.
Missing Key Elements or Using the Wrong Consent Version
Consent can also fail due to documentation and version-control errors, even when the study has been explained properly.
Common examples include:
Missing participant or investigator signatures
Missing dates on the consent form
Use of an outdated or unapproved consent version
Regulations require that only the current, ethics committee approved consent form is used. Even when participant’s understanding is adequate, incorrect or incomplete documentation can lead to serious findings during audits and may invalidate the participant’s consent.
Roles Where Consent Knowledge Is Non-Negotiable
In clinical research, informed consent is not handled by a single role. Different professionals interact with the consent process at different stages of a trial. For some roles, consent knowledge is part of daily responsibilities; for others, it is essential for verification, documentation, and oversight. In all cases, gaps in consent understanding can lead to ethical and compliance issues.
Role
How the Role Interacts with Informed Consent
Clinical Research Coordinator (CRC)
Explains the study to participants, ensures the correct consent version is used, confirms that informed consent is obtained before any study procedure, and manages re-consent when required.
Clinical Research Associate (CRA)
Reviews informed consent forms during monitoring visits, checks signatures, dates, and version control, and verifies that consent was obtained before study-related procedures.
Research Nurse
Supports consent discussions with clinical explanations, identifies participant confusion or concerns, and flags situations where re-consent may be necessary.
Clinical Trial Assistant (CTA)
Maintains and files informed consent documents, tracks approved consent versions, and supports audits and inspections related to consent records.
Why This Matters Across All Roles
Informed consent failures are rarely caused by one individual. A single consent issue often results from multiple small gaps across roles incomplete explanations, missed checks, or documentation errors. When consent knowledge is shared and understood across the team, participant rights are better protected and trial integrity is maintained.
Conclusion
Informed consent is where ethical intent is tested in real clinical practice. It is the point at which regulations, human judgment, communication skills, and accountability intersect. When handled correctly, it protects participants and preserves the credibility of the research. When handled poorly, it exposes trials to ethical failure, regulatory action, and lasting damage to public trust.
For anyone entering clinical research, informed consent is not just a topic to understand, but a responsibility to uphold. It shapes how studies are conducted, how participants are treated, and how confidently a trial can stand up to scrutiny. This is why we at CliniLaunch Research Institute treat this as a foundational competency in our clinical research training programs and not as a procedural checklist.
Learning the informed consent process early builds the mindset required for ethical decision-making, regulatory compliance, and participant-centered research—skills that define competent clinical research professionals across roles and settings.
FAQs
1. What is the informed consent process in clinical research? The informed consent process is an ongoing communication process where participants are informed about a study and voluntarily decide whether to take part.
2. What is the difference between consent and informed consent? Consent is simply agreeing, while informed consent means agreeing after fully understanding the study, risks, benefits, and participant rights.
3. Who is responsible for obtaining informed consent in a clinical trial? Informed consent is usually obtained by the investigator or trained study staff responsible for explaining the study to participants.
4. Is informed consent a one-time signature or an ongoing process? Informed consent is an ongoing process that continues throughout the study, especially when new information or risks arise.
5. What information must be included in an informed consent form? An informed consent form includes study purpose, procedures, risks, benefits, alternatives, privacy details, and the right to withdraw.
6. Can a participant withdraw from a clinical trial after giving consent? Yes, participants can withdraw from a clinical trial at any time without penalty or loss of medical care.
A clinical trial protocol is a structured plan that defines how a clinical study is designed, conducted, monitored, and analyzed. It outlines objectives, participant eligibility, study design, safety measures, and statistical methods to ensure ethical, reliable, and consistent trial execution.
Every clinical trial operates within a clinical trial protocol, even though most beginners only encounter it as a document to be followed. In reality, the protocol is what turns a research idea into a controlled, ethical, and measurable clinical study. Without it, trials would vary from site to site, decisions would be inconsistent, and patient safety would be difficult to protect.
For anyone entering clinical research, understanding how trials are structured is more important than memorizing regulations or job titles. The protocol sits at the center of that structure. It connects scientific objectives with real-world execution and ensures that everyone involved is working from the same plan.
This blog explains what a clinical trial protocol is, why it exists, and how it shapes the way clinical research is planned, conducted, and evaluated in practice.
What Is a Clinical Trial Protocol and Why It Exists
A clinical trial protocol is the written plan that explains how a clinical study will be carried out from start to finish. It defines what the study is trying to answer, who can participate, what procedures will be performed, how safety will be monitored, and how results will be analyzed.
Clinical trial protocols exist because clinical research cannot rely on informal decision-making. Studies involve human participants, medical interventions, and regulatory oversight. The protocol establishes clear rules before the trial begins so that actions taken during the study are consistent, justified, and defensible.
By setting these rules in advance, the protocol serves two critical purposes. First, it protects participants by defining eligibility criteria, visit schedules, and safety assessments. Second, it protects the scientific integrity of the study by ensuring that data is collected and analyzed in a structured and reliable way.
In practice, the clinical trial protocol acts as both a scientific blueprint and an operational guide, making it possible for clinical trials to be ethical, reproducible, and acceptable to regulators.
Who Uses a Clinical Trial Protocol
A clinical trial protocol is used by everyone involved in a clinical study:
Investigators and doctors use it to understand how the study should be conducted and how participants should be treated.
Clinical Research Coordinators (CRCs) follow the protocol to schedule visits, perform procedures, and collect data correctly.
Clinical Research Associates (CRAs) use it to check whether the trial is being conducted according to plan.
Data management and statistics teams rely on the protocol to know what data to collect and how it should be analyzed.
Ethics committees and regulators, such as the FDA and ICH-GCP, review the protocol to ensure the study is ethical, safe, and scientifically sound.
In simple terms, the protocol in clinical trials acts as a shared guidebook for all stakeholders.
What Is Included in a Clinical Trial Protocol?
A clinical trial protocol contains clearly defined sections that explain why a study is conducted, how it will be carried out, and how safety and results will be evaluated. Each section plays a specific role in ensuring that the trial is ethical, consistent, and scientifically reliable.
1. Trial Identification and Administrative Details
This is the identity card of the study. It includes the official study title, protocol number, trial phase, sponsor name, investigator details, and version history.
Why it matters: These details establish investigator responsibilities, trace accountability, and ensure that every site, auditor, and regulator is working from the same approved version. Any mismatch here is a compliance problem, not a clerical error.
2. Background and Scientific Rationale
This section answers a simple but brutal question: Why does this study deserve to exist?
It summarizes current medical knowledge, gaps in evidence, and limitations of existing treatments. The scientific rationale justifies exposing real humans to risk and effort. Without a solid rationale, the study fails both scientifically and ethically.
This is where a clinical trial protocol definition moves beyond theory and proves relevance with data and prior research.
For example, the AURORA cardiovascular outcomes trial was conducted because patients on long-term dialysis had high cardiovascular risk, yet there was insufficient evidence that statins reduced events in this population.
3. Study Objectives and Endpoints
Here, the protocol stops being philosophical and becomes measurable.
Objectives state what the study is trying to prove.
Endpoints define how that proof will be measured.
Primary and secondary endpoints are clearly separated to avoid post-hoc manipulation. This clarity protects the study from biased interpretation and supports regulatory compliance during review.
A weak endpoint definition is one of the fastest ways to kill a study’s credibility.
The study design and methodology section explains:
Trial type (randomized, controlled, open-label, etc.)
Treatment arms and comparators
Randomization and blinding methods
Duration and follow-up structure
Good clinical trial protocol design ensures results are scientifically valid and defensible. Poor design guarantees wasted time, money, and participants.
5. Study Population and Eligibility Criteria
This section defines who gets in and who stays out.
Clear inclusion and exclusion criteria protect participants and prevent noise in the data. They also directly affect how widely the results can be applied in real clinical practice.
Eligibility criteria are a core part of risk benefit assessment. Enrolling the wrong population can expose patients to unnecessary risk or dilute meaningful outcomes.
A well-written schedule ensures consistency across sites and supports accurate data collection and management. Ambiguity here leads to protocol deviations, not flexibility.
7. Safety Monitoring and Risk Management
This section defines how participant safety is actively protected, not just promised.
It explains:
Adverse event reporting
Serious adverse event escalation
Stopping rules and discontinuation criteria
Ongoing safety review processes
This is where ethical considerations in clinical trials meet legal obligation. Continuous safety monitoring is mandatory under global clinical trial protocol guidelines, especially for studies conducted under regulatory frameworks like INDs.
Without rigorous controls, even a perfectly designed trial becomes unusable. Regulators care as much about how data was collected as they do about the results themselves.
9. Statistical Considerations
This is where math prevents false conclusions.
The protocol defines:
Sample size calculation
Statistical tests and assumptions
Power (typically 80–90%)
Significance thresholds
Predefining statistics protects the study from selective analysis and supports transparent interpretation. Changing numbers later is not “optimization”; it’s a red flag.
For example, The WOSCOPS trial used predefined statistical power calculations to ensure sufficient participants were enrolled to detect meaningful treatment effects.
10. Ethical Considerations and Informed Consent
No participant enters a trial without this section being rock solid. The protocol explains the informed consent process, confidentiality safeguards, and participant rights. Consent is not a formality. It is an ongoing ethical obligation backed by global standards like ICH GCP. This section anchors the entire study in human protection, reinforcing that compliance exists to serve people, not paperwork.
Build industry-ready skills to work across real clinical trial environments, from study initiation to close-out. Learn how clinical research actually operates in hospitals, CROs, pharma companies, and research organizations, with a strong focus on compliance, documentation, and trial execution.
In a clinical trial, multiple documents are used at different stages of the study. While the clinical trial protocol sets the overall direction, other documents support communication, execution, analysis, and compliance. Understanding how these documents differ helps clarify who uses what and at which point in the trial.
Protocol vs Informed Consent Form (ICF)
The clinical trial protocol is a technical document created for scientific and regulatory review. It defines the study framework and governs how the trial must be conducted.
The Informed Consent Form exists to support participant decision-making. Its role is ethical rather than operational; it ensures participants understand the study before agreeing to take part.
Protocol vs Investigator Brochure (IB)
The protocol focuses on trial conduct, while the Investigator Brochure focuses on knowledge transfer. The IB equips investigators with background information needed to use the investigational product safely, but it does not dictate how the study itself is running.
Protocol vs Statistical Analysis Plan (SAP)
The protocol establishes the analytical intent of the study on what outcomes matter and why. The SAP translates that intent into executable statistical instructions, ensuring that analysis of decisions is locked before results are examined.
Protocol vs Case Report Forms (CRFs)
The protocol defines what should be observed in a participant. CRFs exist only to capture those observations in a structured, auditable way. If the protocol changes, CRFs must be updated to remain aligned.
Protocol Amendments vs Protocol Deviations
Amendments reflect controlled evolution of the study plan, while deviations represent exceptions that occur during real-world execution. Both are tracked to assess their impact on safety and data integrity under ICH Good Clinical Practice.
How These Documents Function in a Trial
Document
When It Is Used
Primary Owner
What It Enables
Clinical Trial Protocol
Before and throughout the trial
Sponsor
Regulatory approval and trial governance
Informed Consent Form (ICF)
Before participant enrollment
Investigator / IRB
Ethical enrollment of participants
Investigator Brochure (IB)
Before site initiation and during trial
Sponsor
Investigator training and product safety awareness
Statistical Analysis Plan (SAP)
Before database lock
Biostatistics team
Predefined, unbiased data analysis
Case Report Forms (CRFs)
During participant visits
Data management
Standardized data capture
Protocol Amendment
When trial design needs revision
Sponsor
Controlled updates to study conduct
Protocol Deviation
When protocol is not followed
Site / Monitor
Documentation of execution gaps
Understanding how protocols translate into statistical plans and analysis is essential for roles that work closely with trial data and reporting.
Advanced Diploma in
Clinical SAS
Build practical skills in clinical data analysis and reporting using SAS, aligned with regulatory standards used in clinical trials. Learn how clinical trial data is cleaned, analyzed, and presented for regulatory submissions and study reporting.
Common Misconceptions About Clinical Trial Protocols
People new to clinical research often misunderstand what a clinical trial protocol actually does. These misconceptions usually come from seeing the protocol as a static or purely regulatory document, rather than a practical guide used throughout a trial.
1. The protocol is just paperwork
Many believe the protocol exists only to satisfy regulators.
In reality, the protocol guides daily trial activities such as participant visits, safety assessments, dosing decisions, and data collection.
2. Once written, the protocol never changes
It’s commonly assumed that protocols are fixed and cannot be modified.
In practice, protocols can be updated through approved amendments when scientific, operational, or safety-related changes are needed.
3. Only regulators care about the protocol
Another misconception is that the protocol is relevant only during inspections.
In reality, investigators, Clinical Research Coordinators, monitors, data managers, and statisticians rely on the protocol to perform their roles consistently.
Case Study 1: When the Protocol Made the Call
During a clinical trial, a participant developed unexpected safety symptoms after dosing, leaving the site team unsure whether treatment should continue. Instead of relying on judgment, the team followed the clinical trial protocol, which had already defined stopping rules and reporting timelines. Treatment was discontinued and the event was reported as outlined in FDA IND safety reporting requirements.
4. Protocol deviations mean the study has failed
Deviations are often viewed as signs of poor-quality trials.
In real-world settings, deviations are expected. What matters is how they are documented, assessed, and managed.
5. Participants read the clinical trial protocol
Some assume participants are given the full protocol.
In reality, participants interact only with the Informed Consent Form, which explains the study in plain language. The protocol remains a technical document used by the research team.
6. The protocol and SOPs are the same
Protocols and Standard Operating Procedures are often confused.
SOPs describe how an organization operates in general, while the protocol defines how one specific clinical trial must be conducted.
Case Study 2: A Missed Visit That Didn’t Break the Trial
In another study, a participant missed a scheduled visit due to illness, raising concerns about protocol compliance. The team reviewed the protocol, documented the deviation, completed follow-up assessments, and allowed the participant to continue as described in standard clinical study conduct practices outlined by ClinicalTrials.gov.
Conclusion
Clinical trial protocols form the backbone of how clinical research is planned, executed, and evaluated. They bring together scientific intent, participant safety, regulatory expectations, and operational clarity into a single framework that guides decisions throughout the life of a trial.
For anyone building a career in clinical research, protocol knowledge goes beyond understanding procedures; it reflects the ability to think critically, act responsibly, and respond correctly when real-world challenges arise. Strong protocol understanding supports ethical conduct, improves cross-functional collaboration, and ensures consistency across trial sites.
At CliniLaunch Research Institute, we approach protocol knowledge in our clinical research training programs as a critical capability to develop, not just a document to follow. Ultimately, mastering the protocol is what enables clinical research professionals to contribute meaningfully to high-quality, credible research and build sustainable careers in the field. This is why understanding what is clinical trial protocol is foundational for anyone serious about a career in clinical research.
Frequently Asked Questions (FAQs)
1. What is a clinical trial protocol in simple terms?
A clinical trial protocol is a detailed plan that explains how a clinical study will be conducted, including who can participate, what treatment is given, how safety is monitored, and how results are analyzed.
2. Why is a clinical trial protocol required before starting a study?
A protocol is required to ensure the trial is scientifically sound, ethically conducted, and safe for participants. It prevents decisions from being made midway and ensures consistency across all study sites.
3. Who prepares the clinical trial protocol?
Clinical trial protocols are developed collaboratively by sponsors, investigators, statisticians, and regulatory experts to ensure scientific validity, feasibility, and regulatory compliance.
4. Can a clinical trial protocol be changed after the study starts?
Yes. A protocol can be modified through approved protocol amendments if new safety, scientific, or operational information arises. Any change must be reviewed and approved before implementation.
5. What happens if a protocol is not followed?
When a protocol is not followed, it is documented as a protocol deviation. Deviations are reviewed to assess their impact on participant safety and data quality and do not automatically invalidate a study.
6. How is a clinical trial protocol different from informed consent?
The protocol is a technical document used by the research team, while the informed consent form is written for participants to help them understand the study and voluntarily agree to participate.
7. Why is protocol knowledge important for clinical research careers?
Protocol knowledge helps professionals make correct decisions, handle real-world trial situations, and communicate effectively across teams. It is a core skill evaluated in clinical research roles.
8. Do beginners need to understand statistics in a clinical trial protocol?
Yes, at a basic level. Understanding concepts like sample size, endpoints, and statistical power helps professionals understand why trials are designed in a certain way and how results are interpreted.
9. Are clinical trial protocols the same across all studies?
No. While protocols follow standard guidelines such as ICH Good Clinical Practice, each protocol is customized based on the study objective, population, and treatment being evaluated.
Site monitoring in clinical trials is the process of overseeing study conduct at investigator sites to ensure protocol compliance, patient safety, data accuracy, and regulatory compliance. It is primarily carried out by Clinical Research Associates through planned monitoring visits across the trial lifecycle.
Site monitoring in clinical trials is a critical control mechanism that ensures a study is conducted exactly as approved. It exists to confirm that clinical trial activities follow the protocol, meet regulatory compliance requirements, and uphold ethical standards throughout the trial lifecycle.
At the center of this process is the Clinical Research Associate (CRA), who serves as the operational link between the sponsor and the investigator site. Through structured site monitoring visits, the CRA verifies that patient safety is protected, trial data is accurate and traceable, and essential documents are properly maintained.
Errors in trial conduct or documentation, if left undetected, can compromise data integrity, delay regulatory submissions, or raise serious compliance concerns during inspections. For this reason, site monitoring in clinical trials is not a routine formality. It is a safeguard that supports clinical trial oversight and ensures that studies generate reliable and credible results.
This blog explains what site monitoring in clinical trials involves, the different types of clinical trial monitoring used in practice, and the step-by-step site monitoring process followed from site setup to study close-out, supported by a real-world case example.
What is Clinical Trial Monitoring?
Clinical trial monitoring is a systematic process used to ensure that a clinical study is conducted, recorded, and reported in accordance with the approved protocol, Good Clinical Practice (GCP), regulatory requirements, and ethical principles.
In practical terms, clinical trial monitoring functions as an ongoing quality control activity. It focuses on verifying that participant rights and safety are protected, that adverse event reporting is accurate and timely, and that trial data reflects what actually occurred at the site. This includes reviewing source documents, confirming protocol compliance, and ensuring that deviations are identified, documented, and addressed appropriately.
An important aspect of site monitoring in clinical trials is its role in maintaining regulatory inspection readiness. Well-monitored sites are more likely to demonstrate compliance because essential documents, such as the Investigator Site File and Trial Master File, are kept current and complete. Through regular monitoring, sponsors gain confidence that the trial is being conducted as intended and that the data generated can withstand regulatory review.
Types of Clinical Trial Monitoring
Clinical trials differ in complexity, risk profile, geographic spread, and data volume. For this reason, site monitoring in clinical trials is not performed using a single fixed approach. Instead, different types of clinical trial monitoring are applied based on study needs, regulatory expectations, and risk assessment outcomes.
Each monitoring method serves a specific purpose, and in practice, most trials use a combination rather than relying on only one approach.
On-Site Monitoring
On-site monitoring is the traditional and most direct form of site monitoring in clinical trials. In this approach, the Clinical Research Associate conducts monitoring visits by physically visiting the investigator site. These visits allow the CRA to directly observe trial conduct and verify that study procedures are being followed exactly as described in the protocol.
During an on-site monitoring visit, the CRA reviews source documents to perform source data verification, checks informed consent documentation, assesses adverse event reporting, and evaluates drug accountability and storage conditions. Essential documents maintained in the Investigator Site File are also reviewed to confirm regulatory compliance.
Because the CRA is present at the site, on-site monitoring allows for immediate clarification of issues and direct interaction with site staff. However, it is time-intensive and contributes significantly to monitoring-related costs in clinical trials.
Remote Site Monitoring
Remote site monitoring allows the CRA to conduct monitoring activities without physically visiting the site. Instead, monitoring is performed using secure electronic systems such as Electronic Data Capture platforms, electronic Trial Master Files, and Clinical Trial Management Systems.
Through remote monitoring, the CRA can review trial data, track protocol deviations, assess documentation completeness, and follow up on monitoring findings in a timely manner. This approach improves efficiency, reduces travel requirements, and allows more frequent data review compared to traditional on-site visits.
Remote site monitoring is particularly effective for ongoing data checks and document reviews. However, it has limitations when it comes to verifying physical processes, investigational product handling, and site facilities.
Centralized Monitoring
Centralized monitoring is a data-focused approach in which study data from all participating sites is reviewed centrally by the sponsor or contract research organization. Using statistical tools and data analytics, centralized monitoring helps identify trends, outliers, missing data, or unusual patterns that may indicate quality or compliance issues.
This method supports early risk detection across multiple sites and enhances overall clinical trial oversight. Centralized monitoring is especially useful in large, multi-center studies where consistent site-level issues may not be immediately visible through individual monitoring visits.
While centralized monitoring strengthens trial-level oversight, it does not replace site-level verification and is typically used alongside on-site or remote monitoring.
Risk-Based Monitoring (RBM)
Risk-based monitoring is an approach that focuses monitoring efforts on the aspects of a trial that pose the greatest risk to participant safety and data integrity. Instead of applying the same level of monitoring to all sites and activities, RBM uses predefined risk assessments and ongoing data evaluation to guide monitoring intensity.
Under risk-based monitoring, high-risk processes such as informed consent, primary endpoint data, and safety reporting receive greater attention, while lower-risk activities may be monitored less frequently. This approach allows resources to be used more effectively while maintaining regulatory compliance.
RBM typically combines centralized monitoring, remote monitoring, and targeted on-site monitoring as part of a structured site monitoring plan.
Hybrid Monitoring
Hybrid monitoring combines elements of on-site and remote monitoring. In this approach, critical activities such as source data verification, informed consent verification, and drug accountability are performed during on-site visits, while routine data reviews and document checks are handled remotely.
Hybrid monitoring provides a balanced approach, maintaining oversight of high-risk areas while improving efficiency. As clinical trials increasingly adopt digital systems, hybrid monitoring has become a widely used model in modern studies.
Comparison on types of clinical trial monitoring
Monitoring Type
Where It Happens
Key Activities
Strengths
Limitations
On-Site Monitoring
At the clinical trial site
SDV, IP checks, IC review, facility observation
Most comprehensive; direct oversight
Time-consuming; travel cost
Remote Monitoring
Off-site (online review)
EDC review, document checks, communication
Fast, cost-effective, continuous access
Limited ability to verify physical processes
Centralized Monitoring
Sponsor/CRO central systems
Data analytics, trend checks, anomaly detection
Early detection of deviations across sites
Does not replace site-level verification
Risk-Based Monitoring (RBM)
Combination of methods
Risk assessment, targeted checks
Optimizes resources; focuses on critical risks
Requires strong data systems & planning
Hybrid Monitoring
Mix of on-site + remote
Critical tasks on-site, routine tasks remote
Balanced efficiency and quality
Coordination needed between monitoring types
Clinical Trial Monitoring Step-by-Step Process
The site monitoring process in clinical trials follows a structured sequence of visits conducted across the study lifecycle. Each stage serves a distinct purpose, but together they ensure protocol compliance, patient safety monitoring, and data integrity from study start to closure.
The Clinical Research Associate is responsible for planning, executing, documenting, and following up on these monitoring activities as part of ongoing clinical trial oversight.
Pre-Study Visit (PSV): Assessing Site Readiness
The Pre-Study Visit is conducted before a site is authorized to participate in a clinical trial. Its primary objective is to assess whether the site is capable of conducting the study in accordance with the protocol and regulatory requirements.
During the PSV, the CRA evaluates the site’s infrastructure, including clinical facilities, investigational product storage areas, and data handling systems. The qualifications and experience of the investigator and site staff are reviewed to ensure they are appropriate for the study. The CRA also assesses whether the site can manage essential documents, adverse event reporting, and patient records in a compliant manner.
This visit plays a preventive role. By identifying gaps early, the CRA can guide the site on corrective actions before trial initiation, reducing the risk of compliance issues later in the study.
Site Initiation Visit (SIV): Preparing the Site for Trial Conduct
The Site Initiation Visit formally marks the transition from site preparation to active trial conduct. At this stage, the CRA ensures that the site fully understands the study requirements and is ready to enroll participants.
During the SIV, the CRA reviews the approved protocol in detail with the site team, explaining study objectives, eligibility criteria, visit schedules, and safety reporting expectations. Training is provided on informed consent procedures, data entry into the EDC system, and handling of investigational products. The CRA also confirms that all essential documents are in place and that the site monitoring plan is clearly understood.
A well-executed SIV establishes consistency in trial conduct and reduces the likelihood of protocol deviations during enrollment and follow-up.
Routine monitoring visits are conducted at regular intervals throughout the trial and represent the core of site monitoring in clinical trials. These visits allow the CRA to verify that the study continues to be conducted as approved.
During routine monitoring visits, the CRA reviews participant eligibility, confirms that informed consent was obtained correctly, and monitors patient safety through adverse event reporting. Data entered into the CRF or EDC system is compared with source documents as part of source data verification. The CRA also reviews the Investigator Site File to ensure that essential documents remain current and complete.
Any issues identified during these visits are documented as monitoring findings. The CRA works with the site to resolve these issues and, where necessary, supports corrective and preventive actions to prevent recurrence.
Close-Out Visit: Completing Site Responsibilities
The Close-Out Visit is conducted once all trial activities at the site have been completed. The purpose of this visit is to ensure that the site has fulfilled all protocol, regulatory, and documentation requirements before the study is formally closed.
During the close-out visit, the CRA confirms that all data queries have been resolved and that the study data is complete and accurate. Essential documents are reviewed to ensure proper archiving, and regulatory compliance is verified. The CRA also ensures that investigational products are returned or destroyed according to the protocol and applicable regulations.
This final monitoring stage ensures that the site is prepared for audits or inspections and that the study can progress confidently toward analysis and reporting.
Case Study on Clinical Trial Monitoring
How a Routine Monitoring Visit Prevented Data Integrity Issues
This case illustrates how effective site monitoring in clinical trials directly protects data quality and scientific validity.
During a routine monitoring visit, the Clinical Research Associate observed that a study participant scheduled for a Day 5 visit arrived nearly two hours later than planned. Despite the delay, the site staff proceeded with dosing and pharmacokinetic (PK) or pharmacodynamic (PD) sample collection without documenting the deviation or questioning its impact.
At first glance, the situation appeared operationally minor. However, in clinical trials involving PK or PD assessments, sample timing is critical. Even small deviations can significantly affect data interpretation.
Identification of the Issue
When the CRA later reviewed the source documents, a serious discrepancy became evident. The visit was documented as if it had occurred exactly according to the protocol-defined schedule. There was no record of the delayed arrival, no protocol deviation reported, and the PK or PD sample times were recorded based on planned rather than actual collection times.
This meant that the recorded data did not accurately reflect what occurred at the site. Because PK analyses depend on precise timing relative to dosing, the inaccurate documentation had the potential to distort the participant’s concentration profile and compromise the scientific integrity of the dataset.
CRA Action and Follow-Up
Having directly observed the deviation, the CRA escalated the issue to the Principal Investigator and the sponsor. The CRA ensured that the source documents were corrected to reflect the actual visit and sample collection times and that a formal protocol deviation was documented.
In addition, the CRA supported corrective and preventive actions. These included targeted protocol and GCP retraining for the site staff and the introduction of a checklist to reinforce real-time documentation during critical visits. These actions were aimed at preventing similar issues in future visits.
Outcome and Key Learning
Because the issue was identified and addressed promptly, inaccurate PK data was prevented from entering the final analysis. The CRA’s intervention preserved the reliability of the study data and supported regulatory compliance.
This case highlights the value of routine monitoring visits and demonstrates how vigilant site monitoring helps protect patient safety, data integrity, and overall trial credibility. It also reinforces why site monitoring is a critical safeguard rather than a procedural formality.
Conclusion
Site monitoring is one of the most operationally critical functions in clinical trials. It is where protocol design, regulatory expectations, and real-world site execution intersect. Understanding how site monitoring works—across different monitoring types and visit stages—provides a practical view of how clinical trials are actually controlled and safeguarded.
For individuals looking to enter clinical research, this knowledge is not optional. Roles such as Clinical Research Coordinator, Clinical Trial Assistant, and Clinical Research Associate all require a working understanding of site monitoring, protocol compliance, essential documentation, and patient safety oversight.
At CliniLaunch Research Institute, the PG Diploma in Clinical Research is designed to build this exact operational understanding. The program focuses on real clinical trial workflows, including site monitoring processes, CRA responsibilities, regulatory compliance, and inspection readiness—preparing learners to function confidently in entry-level and growing clinical research roles.
For those aiming to move from academic knowledge to industry-ready capability, structured training aligned with real trial operations makes the difference.
Frequently Asked Questions (FAQs)
Who decides how site monitoring will be conducted in a clinical trial?
The sponsor, often with input from the CRO, defines the site monitoring plan. This plan outlines the monitoring approach, visit frequency, and responsibilities based on study risk and complexity.
Is site monitoring mandatory for all clinical trials?
Yes. Some form of site monitoring is required for all interventional clinical trials. The method may vary, but oversight of site activities is always expected by regulators.
Can a clinical trial run without on-site monitoring?
In some studies, monitoring may rely more on remote or centralized methods. However, critical activities such as informed consent and investigational product handling usually still require on-site verification at some stage.
What happens if monitoring issues are not corrected?
Unresolved monitoring findings can lead to protocol deviations, regulatory observations, delayed approvals, or rejection of trial data during inspections.
How are monitoring findings documented?
Monitoring findings are recorded in a monitoring visit report. The site is required to respond, and corrective and preventive actions are tracked until closure.
Does site monitoring replace audits or inspections?
No. Site monitoring is a routine oversight activity. Audits and regulatory inspections are independent reviews conducted by sponsors or authorities to assess overall compliance.
Is site monitoring only the responsibility of the CRA?
The CRA leads monitoring activities, but investigators and site staff are responsible for correcting issues and maintaining compliance at the site.
Why do beginners struggle with site monitoring concepts?
Most academic programs focus on theory, while site monitoring involves operational decision-making, documentation control, and real-time risk management that are learned through practice.
This Blog explores practical biomedical engineering career alternatives that align with how the healthcare and life sciences industry operates today. For many biomedical engineers, career realities differ from expectations set during their academic years. While the degree prepares graduates for innovation. While the degree builds strong foundations in innovation and medical technology, the availability of core roles remains limited across regions. This gap has led many graduates to actively explore alternative careers for biomedical engineers that better align with current industry demand.
Even professionals working in core biomedical roles often experience slow growth, limited specialization, and reduced exposure to high-value areas. At the same time, the healthcare ecosystem is evolving rapidly, driven by digital platforms, data-intensive clinical systems, AI-enabled diagnostics, cloud infrastructure, and stricter regulations. As a result, choosing an alternative career for biomedical engineers has become a practical and sometimes necessary step to remain relevant and future ready.
A 2021 BME Career Exploration study highlights this shift, showing that many graduates now transition into regulated, data-driven, and technology-enabled healthcare roles. These alternative career paths for biomedical engineers leverage core strengths such as systems thinking, analytical ability, and biological understanding, offering clearer growth pathways and long-term career stability. As a result, many graduates now actively consider non-core jobs for biomedical engineers that offer clearer growth, stability, and industry alignment.
The sections below outline how each alternative career for biomedical engineers aligns with current healthcare industry needs, skill requirements, and long-term growth potential. Understanding how different roles evolve over time helps biomedical graduates evaluate long-term biomedical engineers’ career paths beyond traditional assumptions.
Alternative careers for biomedical engineers
The following sections outline structured healthcare careers for biomedical engineers that leverage medical knowledge, regulatory awareness, and system-based thinking.
1. Clinical Research roles
Below are some of the most practical and industry-relevant who want to work beyond traditional core engineering roles while staying connected to healthcare.
Entry-Level Roles You Can Target
Clinical Data Coordinator
Clinical Data Associate
Clinical Trial Assistant (CTA)
Clinical Research Coordinator (CRC)
Pharmacovigilance Associate / Drug Safety Associate
These clinical research roles for biomedical engineers focus on trial execution, data integrity, and regulatory compliance across global studies.
Clinical research focuses on executing and managing clinical trials that test the safety and effectiveness of drugs, devices, and therapies. The work is centered around patient data, documentation, timelines, and regulatory compliance. These roles ensure trials are conducted strictly as per protocol so that results are acceptable to regulators. This is structured, process-driven execution, not discovery research or analytics.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into clinical research because they are comfortable with structured data, medical terminology, and regulated workflows. The roles reward consistency, attention to detail, and protocol adherence rather than innovation or design. For BMEs who want to stay close to healthcare systems and real-world clinical impact, this is a practical and stable career path.
Career Progression, Salary, and Companies
Career progression (typical): Entry-level role → Senior Associate / Analyst → Manager (role-specific) Growth depends on trial exposure, process mastery, and regulatory experience.
Average entry-level salary (India): Most entry-level clinical research roles start between ₹2.5–4.5 LPA, depending on role, organization, and city. CROs generally offer more consistent compensation than hospital-based roles.
Hospitals and academic research centers conducting sponsored trials
Outlook: Clinical research remains stable as regulatory trials continue irrespective of market cycles. With trials becoming more global and data-intensive, the demand for compliant, well-documented execution continues to rise.
How to Get Started
Start by identifying one entry-level role and aligning your preparation toward it rather than applying broadly. Build a clear understanding of the clinical trial lifecycle, GCP principles, and role-specific workflows. For candidates without industry exposure, a structured program like Advance Diploma in Clinical Research helps bridge the gap by providing domain context, practical workflows, and hiring alignment. If internships or site-level opportunities are accessible, they should be pursued alongside or immediately after training. Networking with professionals already working in CROs or trial sites helps clarify expectations early and avoid misaligned roles.
Aspect
Details
Domain
Clinical Research
Core Focus
Trial execution, data integrity, documentation, compliance
Entry-Level Roles
CDM, CTA, CRC, PV, Regulatory, Clinical Ops, Medical Writing
Entry Salary (India)
₹2.5–4.5 LPA (average)
Hiring Organizations
CROs, Pharma, Biotech, Hospitals
Key Skills Needed
GCP basics, process discipline, clinical context
Career Growth
Associate → Analyst → Manager
Long-Term Outlook
Stable, compliance-driven, globally relevant
Advanced Diploma in
Clinical Research
Develop industry-ready clinical research skills used across pharmaceutical companies, CROs, and healthcare organizations. Learn how clinical trials are designed, conducted, monitored, and regulated, while gaining hands-on exposure to real-world clinical research workflows and compliance standards.
Clinical Trial Management, ICH-GCP & Regulatory Compliance, Clinical Data Management Basics, Pharmacovigilance Fundamentals, Trial Documentation & Monitoring, Ethics Committees & Audits, Career Role Readiness (CRA, CTA, CDM)
Other Courses
Clinical SAS
Medical Coding
Biostatistics
2.Medical Coding roles
Entry-Level Roles You Can Target
Medical Coder (ICD-10 / CPT – Trainee / Junior)
Certified Professional Coder (CPC – Entry Level)
Medical Coding Analyst (Junior)
Healthcare Documentation Specialist
Medical Billing & Coding Associate
Revenue Cycle Management (RCM) Associate
Medical coding jobs for biomedical engineers offer a structured, documentation-driven path within healthcare operations. Documentation-driven and compliance-focused roles represent some of the most accessible biomedical engineering jobs outside core engineering functions.
Medical coding focuses on translating clinical documentation such as physician notes, discharge summaries, diagnostic reports, and procedure records into standardized medical codes used for billing, reimbursement, audits, and compliance. The work is documentation-heavy, rule-based, and governed by strict coding guidelines and payer regulations. Accuracy and consistency are critical, as coding errors directly affect revenue, audits, and legal compliance. This is operational healthcare work, not clinical decision-making or biomedical research.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into medical coding because they already understand medical terminology, human anatomy, disease processes, and clinical workflows. The role rewards attention to detail, structured interpretation of medical records, and adherence to classification standards rather than engineering design or innovation. For BMEs who prefer stable, desk-based healthcare roles with clear rules and measurable output, medical coding offers a predictable and scalable career path.
Career Progression, Salary, and Companies
Career progression (typical): Junior Medical Coder → Senior Coder / Coding Analyst → Auditor / Team Lead → Coding Manager / Compliance Specialist.
Growth depends on coding accuracy, certification upgrades, specialty exposure (e.g., inpatient, surgical, risk adjustment), and audit experience.
Average entry-level salary (India): Most entry-level medical coding roles start between ₹2.0–4.0 LPA, depending on certification status, organization, and city. Certified coders generally progress faster than non-certified entrants.
Outlook: Medical coding remains stable due to the ongoing need for standardized billing, insurance processing, and regulatory audits. While automation assists in coding, human coders are still required for complex cases, audits, and compliance-driven reviews, ensuring steady demand.
How to Get Started
Start by deciding whether you want to pursue outpatient, inpatient, or specialty coding instead of treating medical coding as a single generic role. Build strong fundamentals in ICD-10-CM, CPT, and medical documentation standards, as accuracy and guideline interpretation matter more than speed at the entry level. For candidates without prior healthcare operations exposure, a structured program such as a Advanced Diploma in Medical Coding help bridge the gap by providing coding framework clarity, real-world chart interpretation practice, and alignment with hiring expectations. Entry-level production roles or internships are critical to gaining volume-based experience and improving productivity benchmarks. Networking with experienced coders and auditors helps candidates understand certification value, audit expectations, and long-term growth paths early.
Build industry-ready skills in medical coding used across hospitals, healthcare providers, insurance companies, and global healthcare services. Learn to accurately convert medical diagnoses, procedures, and services into standardized codes while ensuring compliance, accuracy, and reimbursement of integrity.
Pharmacovigilance Associate / Drug Safety Associate
Case Processing Associate
Safety Data Associate
Argus Safety / PV Systems Associate (Junior)
Pharmacovigilance Executive
Clinical Safety Coordinator
Pharmacovigilance careers for biomedical engineer’s center on safety monitoring, adverse event reporting, and regulatory compliance.
Pharmacovigilance focuses on monitoring, evaluating, and reporting the safety of drugs, vaccines, and medical products during clinical development and post-marketing use. The work involves adverse event processing, safety data review, and regulatory reporting to global health authorities. These roles ensure that safety risks are identified, documented, and communicated accurately across the product lifecycle. This is compliance-driven safety surveillance, not laboratory research or clinical decision-making.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into pharmacovigilance because they are familiar with medical terminology, disease mechanisms, and structured documentation. The role requires careful interpretation of clinical narratives, attention to detail, and adherence to regulatory standards rather than engineering design or experimentation. For BMEs who prefer analytical, documentation-focused healthcare roles with regulatory relevance, drug safety offers a stable and well-defined career path.
Career Progression, Salary, and Companies
Career progression (typical): Drug Safety Associate → Senior Safety Associate → Safety Scientist / PV Lead → Safety Manager
Growth depends on case complexity exposure, regulatory knowledge, safety database experience, and therapeutic area specialization.
Average entry-level salary (India): Most entry-level pharmacovigilance roles start between ₹2.5–5.0 LPA, depending on organization, role scope, and city.
Companies you can apply to:
CROs: IQVIA, ICON, Parexel
Pharma / Biotech companies with in-house safety teams
Pharmacovigilance service providers
Outlook: Pharmacovigilance remains stable due to increasing regulatory scrutiny and global safety reporting requirements. Demand continues if drugs and vaccines remain in use worldwide.
How to Get Started
Begin by understanding core pharmacovigilance workflows such as adverse event reporting, case processing, MedDRA coding, and global regulatory requirements. Candidates should align their preparation toward specific entry-level roles rather than applying broadly. For those without prior industry exposure, a structured program like the Advanced Diploma in Clinical Research, where pharmacovigilance and drug safety are covered as a dedicated module, helps bridge the gap by providing regulatory context and practical workflow understanding. Entry-level roles in CROs or safety teams provide the exposure needed to build accuracy and long-term role clarity.
Aspect
Details
Domain
Pharmacovigilance & Drug Safety
Core Focus
Safety monitoring, case processing, regulatory reporting
Entry-Level Roles
Drug Safety Associate, PV Executive, Case Processor
Entry Salary (India)
₹2.5–5.0 LPA (average)
Hiring Organizations
CROs, Pharmaceutical companies, PV service providers
Regulatory affairs jobs after biomedical engineering focus on compliance, submissions, and interaction with health authorities.
Regulatory affairs focus on ensuring that drugs, medical devices, and clinical studies comply with national and international regulatory requirements. The work involves preparing, reviewing, and maintaining regulatory documents, coordinating submissions, tracking approvals, and supporting interactions with health authorities. These roles ensure products and trials meet defined regulatory standards before and after approval. This is documentation- and compliance-driven work, not laboratory research or product development.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into regulatory affairs because they understand medical concepts, product lifecycles, and structured documentation requirements. The role rewards attention to detail, interpretation of guidelines, and consistency in regulatory communication rather than innovation or experimentation. For BMEs who prefer policy-aligned, process-oriented roles that influence product approvals and compliance, regulatory affairs offer a clear and stable career path.
Outlook: Regulatory affairs remain stable as approval requirements continue to expand globally. Increasing regulatory complexity across regions sustains demand for trained regulatory professionals.
How to Get Started
Start by understanding regulatory fundamentals such as submission types, approval pathways, and global guidelines governing drugs and medical devices. Candidates should prepare specific entry-level roles rather than applying broadly across regulatory functions. For those without industry exposure, a structured program like the Advanced Diploma in Clinical Research, which includes regulatory affairs as a core module, helps build regulatory context, documentation familiarity, and workflow understanding. Entry-level roles in pharma companies or CRO regulatory teams provide practical exposure to submissions and compliance processes. Early networking with regulatory professionals helps clarify specialization paths and expectations.
Biomedical engineers fit well into quality assurance because they are trained to work with structured processes, technical documentation, and regulated environments. The role values attention to detail, risk awareness, and consistency in following procedures rather than innovation or design work. For BMEs who prefer governance-oriented roles that influence compliance and operational quality across healthcare systems, QA offers a stable and well-defined career option.
Career Progression, Salary, and Companies
Career progression (typical): QA Associate → Senior QA Associate → QA Manager → Quality Lead / Head
Growth depends on audit exposure, regulatory knowledge, SOP management experience, and cross-functional coordination.
Average entry-level salary (India): Most entry-level quality assurance roles start between ₹3.0–5.0 LPA, depending on organization, domain, and city.
Companies you can apply to:
CROs: IQVIA, Parexel
Pharma & Biotech companies
Medical device manufacturers
Hospitals and research organizations with QA teams
Outlook: Quality assurance remains stable as regulatory inspections; audits, and compliance requirements continue to intensify globally. Demand persists across clinical research, pharma, and healthcare operations.
How to Get Started
Begin by understanding quality fundamentals such as SOPs, GxP guidelines, deviation management, and audit processes. Candidates should align their preparation toward entry-level QA roles rather than applying broadly across departments. For those without industry exposure, a structured program like the Advanced Diploma in Clinical Research, which includes quality assurance as a core module, helps build compliance context, documentation familiarity, and workflow understanding. Entry-level roles in CROs or regulated organizations provide practical exposure to audits and quality systems. Early interaction with QA professionals helps clarify long-term specialization paths.
Aspect
Details
Domain
Quality Assurance
Core Focus
Compliance, audits, SOP adherence
Entry-Level Roles
QA Associate, Compliance Executive
Entry Salary (India)
₹3.0–5.0 LPA (average)
Hiring Organizations
CROs, Pharma, Medical Devices, Hospitals
Key Skills Needed
Documentation, process discipline, GxP basics
Career Growth
Associate → Manager → Lead
Long-Term Outlook
Stable, audit-driven, globally relevant
6.Health Informatics roles
Entry-Level Roles You Can Target
Health Informatics Associate
Clinical Informatics Coordinator
Healthcare Data Analyst (Junior)
EHR / EMR Support Analyst
Health Information Management (HIM) Executive
Clinical Systems Support Associate
A health informatics career path combines healthcare data, clinical systems, and technology-enabled workflows.
Health informatics focuses on managing, analyzing, and optimizing healthcare data generated from clinical systems such as electronic health records, hospital information systems, and clinical databases. The work involves data accuracy, system workflows, interoperability, reporting, and supporting clinicians and administrators in using health data effectively. These roles sit at the intersection of healthcare, IT systems, and data governance. This is systems and data coordination work, not clinical practice or biomedical research.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into health informatics because they understand clinical workflows, medical terminology, and system-based thinking. The role requires interpreting healthcare data, working with structured systems, and supporting technology-driven care delivery rather than engineering design or experimentation. For BMEs who want to work with healthcare data and digital systems while staying close to clinical environments, health informatics offers a practical and evolving career path.
Career Progression, Salary, and Companies
Career progression (typical): Health Informatics Associate → Informatics Analyst → Senior Analyst / Consultant → Informatics Manager
Growth depends on system exposure, healthcare domain knowledge, data handling skills, and familiarity with clinical information systems.
Average entry-level salary (India): Most entry-level health informatics roles start between ₹3.0–5.5 LPA, depending on organization, system complexity, and city.
Companies you can apply to:
Hospitals and healthcare networks using digital health systems
CROs and pharma organizations with informatics teams
Outlook: Health informatics continues to grow as healthcare systems digitize, and data-driven care becomes standard. Demand increases with expanding EHR adoption, interoperability requirements, and healthcare analytics needs.
How to Get Started
Begin by understanding healthcare data flows, EHR concepts, clinical documentation standards, and basic health data governance. Candidates should target specific entry-level informatics or clinical systems roles rather than applying broadly across IT or analytics positions. At CliniLaunch Research Institute, relevant programs such as the PG Diploma in AI/ML in Healthcare and clinical research programs that expose learners to healthcare data systems and clinical workflows help build foundational informatics understanding. Entry-level roles in hospitals, health IT teams, or healthcare analytics firms provide practical exposure to real-world systems and data environments.
Aspect
Details
Domain
Health Informatics
Core Focus
Healthcare data systems, EHRs, clinical workflows
Entry-Level Roles
Informatics Associate, EHR Analyst
Entry Salary (India)
₹3.0–5.5 LPA (average)
Hiring Organizations
Hospitals, Health IT firms, Analytics companies
Key Skills Needed
Clinical data understanding, systems thinking
Career Growth
Associate → Analyst → Manager
Long-Term Outlook
Growing, data-driven, healthcare-focused
7. Digital Health roles
Entry-Level Roles You Can Target
Digital Health Executive
Health Technology Associate
Clinical Digital Operations Associate
Digital Health Project Coordinator
Healthcare Technology Support Analyst
Digital Health Data Associate (Junior)
Digital health jobs for biomedical engineers involve supporting technology platforms used in modern healthcare delivery.
Digital health focuses on the use of technology to improve healthcare delivery, patient monitoring, clinical workflows, and health data management. The work involves supporting digital platforms such as telemedicine systems, remote patient monitoring tools, clinical dashboards, and healthcare applications. These roles bridge healthcare operations and technology by ensuring digital tools are implemented, used, and maintained effectively. This is technology-enabled healthcare operations, not software development or clinical practice.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into digital health because they understand both healthcare systems and technology-driven workflows. The role values systems thinking, clinical context awareness, and the ability to work across medical and technical teams rather than pure coding or engineering design. For BMEs who want to be part of healthcare innovation without moving into core IT development roles, digital health offers a flexible and future-facing career path.
Career Progression, Salary, and Companies
Career progression (typical): Digital Health Associate → Digital Health Analyst → Senior Analyst / Consultant → Digital Health Manager
Growth depends on platform exposure, healthcare domain knowledge, data handling skills, and cross-functional coordination.
Average entry-level salary (India): Most entry-level digital health roles start between ₹3.0–6.0 LPA, depending on organization, role scope, and city.
Companies you can apply to:
Digital health and health-tech companies
Hospitals adopting telemedicine and digital care platforms
Healthcare analytics and technology consulting firms
Pharma and CROs implementing digital trial solutions
Outlook: Digital health continues to expand as healthcare systems adopt virtual care, remote monitoring, and data-driven decision-making. Demand is driven by technology adoption rather than traditional healthcare hiring cycles.
How to Get Started
Begin by understanding digital health fundamentals such as telemedicine workflows, healthcare data integration, digital clinical platforms, and patient engagement systems. Candidates should target entry-level digital health or healthcare technology coordination roles rather than generic IT positions. At CliniLaunch Research Institute, programs such as the PG Diploma in AI/ML in Healthcare and clinical research programs provide exposure to healthcare data, digital tools, and clinical workflows that are directly relevant to digital health roles. This combination helps learners understand how technology, data, and clinical operations intersect in real healthcare environments. Entry-level roles in health-tech companies or hospital digital teams provide practical exposure to digital healthcare systems.
Aspect
Details
Domain
Digital Health
Core Focus
Healthcare technology, digital platforms, workflows
Healthcare context, systems thinking, coordination
Career Growth
Associate → Analyst → Manager
Long-Term Outlook
Growing, technology-driven, healthcare-focused
8. Data Analytics roles
Data Analytics roles
Data Analyst (Junior)
Healthcare Data Analyst
Clinical Data Analyst (Non-statistical)
Business / Reporting Analyst (Healthcare)
Data Operations Associate
Analytics Support Associate
Healthcare data analytics careers focus on turning clinical and operational data into actionable insights.
Data analytics focuses on collecting, cleaning, analyzing, and interpreting structured data to support decision-making. In healthcare and life sciences, this includes clinical data, operational metrics, patient outcomes, and business performance data. The work involves dashboards, reports, trend analysis, and data validation rather than predictive modeling or advanced research. These roles support operational and strategic decisions using existing data systems. This is applied data analysis, not core data science or algorithm development.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into data analytics because they are comfortable working with data, structured problem-solving, and interpreting technical information within a healthcare context. The role values analytical thinking, logical reasoning, and the ability to translate data into meaningful insights rather than deep programming or mathematical research. For BMEs who want to work with data while staying connected to healthcare and life sciences, data analytics offers a flexible and transferable career option.
Career Progression, Salary, and Companies
Career progression (typical): Junior Data Analyst → Data Analyst → Senior Analyst → Analytics Manager / Lead
Growth depends on domain expertise, data handling skills, reporting accuracy, and exposure to real business or clinical datasets.
Average entry-level salary (India):
Most entry-level data analytics roles start between ₹3.0–6.0 LPA, depending on industry, tools used, and organization.
Companies you can apply to:
Healthcare and life sciences organizations
Analytics and consulting firms
Hospitals and health-tech companies
CROs and pharma companies using data-driven operations
Outlook: Data analytics remain in demand as organizations rely increasingly on data for operational efficiency and decision-making. In healthcare, demand continues due to growing data volumes and digital system adoption.
How to Get Started
Begin by understanding data analytics fundamentals such as data cleaning, basic statistics, reporting, and visualization. Candidates should focus on entry-level analyst roles rather than advanced data science positions. At CliniLaunch Research Institute, programs like the PG Diploma in AI/ML in Healthcare provide exposure to healthcare datasets, analytical tools, and applied use cases that are relevant to analytics roles. This foundation helps learners connect data analysis with real healthcare and clinical scenarios. Entry-level analysts or reporting roles provide the practical experience needed to grow within the analytics domain.
How to Get Started
Begin by understanding data analytics fundamentals such as data cleaning, basic statistics, reporting, and visualization. Candidates should focus on entry-level analyst roles rather than advanced data science positions. At CliniLaunch Research Institute, programs like the PG Diploma in AI/ML in Healthcare provide exposure to healthcare datasets, analytical tools, and applied use cases that are relevant to analytics roles. This foundation helps learners connect data analysis with real healthcare and clinical scenarios. Entry-level analysts or reporting roles provide the practical experience needed to grow within the analytics domain.
Aspect
Details
Domain
Data Analytics
Core Focus
Data analysis, reporting, decision support
Entry-Level Roles
Data Analyst, Reporting Analyst
Entry Salary (India)
₹3.0–6.0 LPA (average)
Hiring Organizations
Healthcare firms, Analytics companies
Key Skills Needed
Data handling, analytical thinking, tools
Career Growth
Analyst → Senior Analyst → Manager
Long-Term Outlook
Stable, data-driven, cross-industry
9. Data Science roles
Entry-Level Roles You Can Target
Data Scientist (Junior)
Associate Data Scientist
Machine Learning Analyst (Entry-Level)
Healthcare Data Scientist (Junior)
AI/ML Analyst (Trainee)
Applied Analytics Associate
Data science represents one of the most advanced life sciences career options for biomedical engineers with analytical strengths.
Data science focuses on using data to build predictive models, identify patterns, and support complex decision-making. In healthcare and life sciences, this includes working with clinical data, patient outcomes, operational datasets, and real-world evidence to generate insights using statistical methods and machine learning techniques. The work involves data preparation, model development, validation, and interpretation rather than routine reporting. This is applied to modeling and analytics, not pure software engineering or academic research.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into data science because they combine analytical thinking with a strong domain understanding of healthcare and biological systems. The role benefits from problem-solving ability, comfort with data-driven reasoning, and the capacity to interpret results within a medical or clinical context. While additional skills in programming and statistics are required, BMEs often adapt well because they already understand the complexity and variability of healthcare data. For those willing to build deeper analytical expertise, data science offers high-impact roles across healthcare and life sciences.
Career Progression, Salary, and Companies
Career progression (typical): Junior Data Scientist → Data Scientist → Senior Data Scientist → Data Science Lead / Manager
Growth depends on model-building capability, domain expertise, problem complexity handled, and business or clinical impact of solutions.
Average entry-level salary (India): Most entry-level data science roles start between ₹4.5–8.0 LPA, depending on skill depth, industry, and organization.
Companies you can apply to:
Healthcare and life sciences analytics firms
Health-tech and AI-driven healthcare companies
Pharma, biotech, and CROs using advanced analytics
Consulting and data science service organizations
Outlook: Data science continues to grow as healthcare organizations adopt AI-driven decision-making and predictive analytics. Demand remains strong for professionals who can combine technical modeling skills with healthcare domain understanding.
How to Get Started
Begin by building strong fundamentals in data handling, statistics, and programming before moving into machine learning concepts. Candidates should target junior or associate data science roles rather than expecting direct entry into advanced modeling positions. At CliniLaunch Research Institute, the PG Diploma in AI/ML in Healthcare provides exposure to healthcare datasets, applied machine learning workflows, and real-world use cases relevant to data science roles. This foundation helps learners connect algorithms with clinical and healthcare problems. Entry-level analytics or ML trainee roles provide the practical experience needed to progress within the data science domain.
Aspect
Details
Domain
Data Analytics
Core Focus
Data analysis, reporting, decision support
Entry-Level Roles
Data Analyst, Reporting Analyst
Entry Salary (India)
₹3.0–6.0 LPA (average)
Hiring Organizations
Healthcare firms, Analytics companies
Key Skills Needed
Data handling, analytical thinking, tools
Career Growth
Analyst → Senior Analyst → Manager
Long-Term Outlook
Stable, data-driven, cross-industry
10. AI & ML in Healthcare
Entry-Level Roles You Can Target
AI/ML Analyst (Healthcare – Junior)
Healthcare Machine Learning Associate
Clinical AI Analyst
Healthcare Data Science Associate (AI-focused)
AI Solutions Analyst (Healthcare)
Applied AI Analyst (Life Sciences)
AI and ML in healthcare careers apply machine learning models to clinical, imaging, and healthcare datasets.
AI and ML in healthcare focus on applying machine learning models and data-driven algorithms to healthcare, clinical, and life sciences data. The work involves developing, testing, and validating models for use cases such as disease prediction, patient risk stratification, medical imaging support, clinical decision support, and operational optimization. These roles sit at the intersection of healthcare data, analytics, and applied machine learning. This is applied to AI work, not software engineering or academic research.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into AI and ML roles because they understand healthcare data complexity, clinical context, and biological variability. The role requires analytical thinking, problem formulation, and the ability to interpret model outputs in a medical or clinical setting rather than only focusing on algorithms. For BMEs willing to build strong foundations in data handling, statistics, and machine learning, AI and ML in healthcare offer high-impact and future-facing career opportunities.
Career Progression, Salary, and Companies
Career progression (typical): AI/ML Analyst → Machine Learning Engineer / Data Scientist → Senior AI Specialist → AI Lead / Manager.
Growth depends on model deployment exposure, domain-specific use cases handled, and the ability to translate AI outputs into healthcare decisions.
Average entry-level salary (India): Most entry-level AI and ML healthcare roles start between ₹5.0–9.0 LPA, depending on skill depth, tools proficiency, and organization.
Pharma, biotech, and CROs using AI for trials and RWE
Healthcare analytics and AI consulting firms
Hospitals and research organizations adopting AI solutions
Outlook: AI and ML adoption in healthcare continues to expand, driven by increasing data availability and demand for predictive, automated decision-support systems. Roles favor professionals who combine technical skills with healthcare domain understanding.
How to Get Started
Begin by building strong foundations in data analytics, statistics, and programming before moving into machine learning concepts and healthcare use cases. Candidates should target junior or associate AI/ML roles rather than advanced research positions initially. At CliniLaunch Research Institute, the PG Diploma in AI/ML in Healthcare provides structured exposure to healthcare datasets, applied machine learning workflows, and real-world clinical use cases. This helps learners understand how AI models are built, validated, and interpreted within healthcare environments. Entry-level analysts or AI trainee roles provide the practical experience required to progress in this domain.
Aspect
Details
Domain
AI & ML in Healthcare
Core Focus
Applied machine learning, healthcare data modeling
Machine learning fundamentals, data handling, healthcare context
Career Growth
Analyst → Specialist → Lead
Long-Term Outlook
Growing, skill-driven, high-impact
PG Diploma in
AI & ML in Healthcare
Build future-ready skills at the intersection of artificial intelligence and healthcare. Learn how AI and machine learning are applied in clinical research, medical imaging, diagnostics, drug discovery, and healthcare data analytics to solve real-world healthcare problems.
Python for AI & ML, Healthcare Data Analysis, Machine Learning Algorithms, Deep Learning Fundamentals, AI in Medical Imaging & Diagnostics, Predictive Analytics in Healthcare, Real-world Healthcare AI Projects
11. Medical Devices & Application Specialist Roles
Entry-Level Roles You Can Target
Application Specialist (Medical Devices – Junior)
Clinical Application Executive
Product Support Specialist (Medical Devices)
Field Application Associate
Technical Clinical Support Executive
Device Training & Support Associate
Medical device application specialist jobs combine clinical exposure with hands-on device support and training.
Medical device application roles focus on supporting the clinical use, setup, and optimization of medical devices used in hospitals and diagnostic settings. The work involves product demonstrations, user training, troubleshooting, clinical workflow support, and coordination between clinicians and device companies. These roles ensure devices are used safely, effectively, and as intended in real-world healthcare environments. This is application and clinical support work, not device design or core R&D.
Why Biomedical Engineers Fit Well
Biomedical engineers fit well into application specialist roles because they understand medical devices, clinical environments, and technology–user interaction. The role values product knowledge, communication with clinicians, and practical problem-solving rather than engineering design or laboratory research. For BMEs who prefer hands-on clinical exposure and interaction with healthcare professionals, this path offers a direct connection to patient care through technology.
Career Progression, Salary, and Companies
Career progression (typical): Application Specialist → Senior Application Specialist → Product / Clinical Manager → Regional Product Lead
Growth depends on device expertise, clinical exposure, communication skills, and territory or product responsibility.
Average entry-level salary (India): Most entry-level application specialist roles start between ₹3.0–6.0 LPA, depending on device category, organization, and city.
Medical equipment distributors and service partners
Hospitals using advanced medical devices
Outlook: Medical device application roles remain steady as hospitals continue adopting advanced technologies. Demand grows with the introduction of new devices that require structured clinical training and support.
How to Get Started
Begin by understanding basic medical device principles, clinical workflows, and user training requirements. Candidates should target application or clinical support roles rather than pure sales positions. At CliniLaunch Research Institute, exposure gained through the Advanced Diploma in Clinical Research helps learners understand clinical environments, regulatory expectations, and device usage within trials and healthcare settings. Entry-level roles with device companies or distributors to provide hands-on exposure to products and clinical users.
Aspect
Details
Domain
Medical Devices & Applications
Core Focus
Device usage, clinical support, user training
Entry-Level Roles
Application Specialist, Clinical Support
Entry Salary (India)
₹3.0–6.0 LPA (average)
Hiring Organizations
Medical device companies, Hospitals, Distributors
Key Skills Needed
Device knowledge, clinical communication
Career Growth
Associate → Specialist → Manager
Long-Term Outlook
Stable, technology-driven, clinically relevant
Conclusion
Identifying the best careers after biomedical engineering requires clarity on industry expectations, role realities, and long-term skill relevance. For those navigating uncertainty after graduation or early in their careers, selecting an alternative career for biomedical engineers should be a structured decision based on role clarity, industry demand, and long-term relevance rather than short-term trends.
Biomedical engineers, career growth today depends less on job titles and more on how well their skills align with evolving industry needs. Healthcare organizations increasingly seek professionals who understand systems, processes, data, and regulatory expectations. This creates meaningful opportunities beyond traditional roles for those willing to adapt and upskill with clarity. Making informed career choices, understanding role expectations early, and preparing with the right foundation can help biomedical engineers build stable and relevant careers in a changing healthcare landscape.
What is clinical data management is a common question in clinical research, especially when trials generate large volumes of patient data across multiple sites, systems, and teams over long timelines. If this data is not collected and reviewed in a controlled way, even a well-designed study can produce unreliable results, making clinical data management in clinical trials essential for reliable outcomes.
Clinical Data Management exists to prevent this risk by defining how clinical trial data is captured, checked, corrected, stored, and prepared for analysis and regulatory review. Without a structured data management process, trial results cannot be trusted, and regulatory approval becomes uncertain, highlighting the growing importance of clinical data management in clinical research.
Clinical Data Management is important because clinical trial results are only as reliable as the data behind them.
It prevents inconsistent data entry, unresolved discrepancies, and safety data mismatches, ensuring trial data remains accurate, traceable, and acceptable for regulatory review.
What Is Clinical Data Management?
Clinical Data Management (CDM) is the process of handling clinical trial data so that it is accurate, complete, and usable. It covers how patient data is collected, checked, corrected, stored, and finalized during a clinical study.
In a clinical trial, patient information such as medical history, lab results, treatment details, and safety events is recorded at different study sites and entered electronic systems. CDM ensures this information is captured in a consistent format, reviewed for errors or missing values, corrected when needed, and documented properly. By the end of the trial, CDM delivers a clean and finalized database that accurately represents what happened during the study and is ready for analysis.
Why Clinical Trials Depend on Clinical Data Management
Clinical trials depend on clinical data management because trial results are only as reliable as the data used to produce them. Even a scientifically sound study can fail if the underlying data is incomplete, inconsistent, or poorly documented highlighting the importance of clinical data management.
Independent audits of clinical research data have shown that, without rigorous data management controls, datasets can contain anywhere from 2 to as high as 2,784 errors per 10,000 data fields, making it impossible to trust results without systematic data review. Without clinical data management, there is no reliable way to confirm that the collected data accurately reflects what occurred during the trial.
In real clinical trials, patient data is generated across multiple hospitals, investigators, laboratories, and external systems, often over long study durations. Data is entered by different teams, reviewed at different times, and updated as patients progress through the study. Without a structured data management process, discrepancies accumulate, safety information may not align across systems, and missing data goes unnoticed until late in the trial, causing delays and rework.
Clinical data management exists to control these risks. CDM teams ensure that data follows consistent definitions, validation rules, and review processes across all sites and sources. They identify errors early, manage queries with study sites, reconcile safety data, and maintain audit trails for every data change. This prevents data quality issues from reaching the analysis stage and protects the integrity of trial outcomes.
Advanced Diploma in
Clinical Research
Master end-to-end clinical trial management, from site monitoring and patient recruitment to regulatory documentation. This program provides hands-on training with industry-standard tools like EDC systems, CTMS, and eTMF, preparing you for immediate roles in CROs and Pharma.
Why Clinical Data Management Is Critical for Regulatory Approval
Regulatory authorities do not approve clinical trials based on positive outcomes alone. Approval depends on whether the submitted data is accurate, consistent, and transparently managed. Clinical data management ensures this by aligning trial execution with clinical data management guidelines and regulatory expectations.
From a regulator’s perspective, unreliable data invalidates conclusions. CDM ensures that every data point can be traced, reviewed, and explained, supporting ICH GCP compliance throughout the study. Clinical data management ensures that every data point submitted can be explained, verified, and traced back to its source, which is a fundamental expectation during regulatory review
Role of Clinical Data Management Teams in Ensuring Data Quality
Regulatory approval depends on the accuracy, completeness, and traceability of clinical trial data, and this responsibility sits directly with clinical data management teams. Clinical Data Managers oversee how data is collected, reviewed, and corrected across the trial, ensuring it aligns with the study protocol and regulatory requirements. They define review strategies, oversee query resolution, and monitor data quality throughout the study lifecycle.
Data Coordinators and Data Reviewers support this process by continuously checking patient records, laboratory results, and safety data for inconsistencies or missing information. Issues are identified early and resolved with trial sites before they escalate into submission delays or inspection findings. This continuous oversight is what keeps trial data consistent and defensible. These reflect evolving clinical data manager roles and responsibilities.
Role of CDM in Audit Readiness and Regulatory Submission
Complete audit trail documentation is critical during inspections. Clinical data management is central to audit readiness. Clinical Programmers and database-focused CDM professionals maintain validated data systems with complete audit trails that record every data change, including who made the change, when it was made, and why. During regulatory inspections, this traceability is not optional; it is scrutinized in detail.
Clinical programmers and database-focused CDM professionals maintain validated systems with a complete audit trail, recording who changed data, when, and why. This level of traceability is essential during inspections.
CDM teams also prepare clean, standardized datasets that are ready for statistical analysis and regulatory submission. These datasets must follow industry standards and be supported by complete documentation, allowing regulators to review trial data efficiently and confidently. Maintaining this level of control throughout the study supports inspection of readiness and aligns with ICH GCP expectations across the entire clinical trial lifecycle. These practices align with global clinical data management guidelines.
Supporting CDM Roles in Complex or Global Trials
In large or complex clinical trials, additional specialized roles strengthen data management and regulatory preparedness. Clinical Database Designers ensure that study databases are built correctly from the start, aligning data structures with the protocol and submission standards. Data Validation and Standards specialists focus on programmed checks and compliance with required industry formats, reducing the risk of submission of rework.
These roles exist for one reason: to prevent data quality issues from surfacing during regulatory review, when fixes are costly, time-consuming, and sometimes impossible.
Phases of Clinical Data Management
Clinical Data Management runs across the entire study, forming the complete clinical data management lifecycle. These stages together define the clinical data managementprocess used in real-world trials
Clinical Data Management does not happen at the end of a clinical trial. It runs alongside the study from planning to final submission, adapting its focus as the trial progresses. The objective remains constant throughout: ensure that clinical trial data is accurate, consistent, and ready for regulatory review.
In real-world clinical trials, CDM activities are structured across three main phases: Study Start-Up, Study Conduct, and Study Close-Out. Each phase controls a different category of data risk and prepares the study for the next stage of execution or review. Understanding these phases explains how CDM works in practice, not just in theory.
Study Start-Up Phase: Building the Data Foundation
The study’s start-up phase focuses on defining how trial data will be collected and controlled before the first patient is enrolled. Decisions made at this stage determine whether the trial will generate clean, usable data or struggle with inconsistencies for its entire duration.
During start-up, CDM teams translate protocol requirements into a case report form and design the database. The data management plan defines how data will be collected, reviewed, validated, and locked. These activities rely on specialized clinical data management tools. A well-designed database also supports data privacy and security across trial systems.
Common platforms include Medidata Rave, Oracle Clinical, Veeva Vault EDC, and OpenClinica—each an electronic data capture solution aligned with CDISC standards. Validation rules, data standards, and workflows are defined early, so that data is captured consistently across all sites from day one. Weak planning at this stage often leads to extensive rework, delayed timelines, and data quality issues that are difficult or expensive to fix later.
Common tools used in this phase include Electronic Data Capture (EDC) platforms such as Medidata Rave. It is a widely used electronic data capture platform in clinical trials, Oracle Clinical, Veeva Vault EDC, and OpenClinica. Industry data standards like CDISC are also applied early to ensure submission of readiness.
Study Conduct Phase: Controlling Data While the Trial Is Live
Following clinical data management best practices reduces downstream risk.
Once enrollment begins, CDM teams control data in real time, applying clinical data management to best practices. Data is collected from sites and labs, enabling source data verification and ongoing review.
During study conduct, CDM teams perform query management, reconciliation of patient safety data, application of medical coding, and continuous data validation checks, supporting effective data cleaning in clinical trials and maintaining clinical trial data quality. Effective query management prevents delays. The goal is to prevent data issues from accumulating and to ensure that safety and efficacy data remain aligned across systems throughout the trial.
This phase is critical because unresolved discrepancies, inconsistent safety reporting, or delayed data review can directly impact analysis of timelines and regulatory readiness. This supports ongoing data cleaning in clinical trials. All these platforms together function as a clinical data management system.
Common tools used in this phase include EDC query management modules, safety databases such as Argus, medical coding dictionaries like MedDRA and WHO-DD, and built-in reporting dashboards used to monitor data quality and study progress.
Study Close-Out Phase: Finalizing Data for Analysis and Submission
The close-out phase focuses on final reviews and database locks, after which data becomes final for analysis. Tools such as SAS and Pinnacle 21 validate submission of readiness and ensure standards of compliance. At this stage, data changes become highly restricted, making unresolved issues particularly risky.
CDM teams perform final data reviews, confirm that all queries are resolved, verify safety reconciliation, and complete final validation checks. This includes systematic data validation checks. Once these activities are complete, the database is locked. After database lock, the data is considered final and is used for statistical analysis and regulatory submission. Errors discovered after this point often result in delays, additional scrutiny, or challenges during regulatory review.
Common tools used in this phase include statistical and validation tools such as SAS for data consistency checks and Pinnacle 21 for validating submission-ready datasets against CDISC standards.
Why These Phases Matter
Each phase of clinical data management exists to control a specific type of risk. Study start-up prevents structural data issues; study conduct prevents uncontrolled data drift, and study close-out ensures regulatory confidence in the final dataset. Skipping rigor in any phase does not just create operational problems; it directly threatens trial timelines, data credibility, and regulatory approval.
CDM Phase
Primary Focus
What CDM Controls at This Stage
Typical Tools Involved
Study Start-Up
Planning and setup before enrollment
Defines what data is collected, how it is captured, and how it will be validated to avoid structural data issues later
EDC systems (Medidata Rave, Oracle Clinical, Veeva Vault EDC, OpenClinica), CDISC standards
Study Conduct
Ongoing data monitoring during the trial
Ensures data completeness, consistency, and alignment across sites and systems while patients are active
Confirms all data is accurate, resolved, validated, and locked for regulatory use
SAS, Pinnacle 21, submission validation tools
Skills Needed for Clinical Data Management (CDM)
Clinical Data Management is not just about knowing tools or following checklists. CDM professionals balance technical execution with regulatory discipline. Understanding clinical data manager roles and responsibilities is key to career progression. CDM professionals sit at the intersection of trial execution, data integrity, and inspection readiness, which is why their skill set must balance technical execution, process awareness, and regulatory discipline.
Core Technical Skills
Data Quality Review Maintaining clinical trial data quality is the primary goal. CDM professionals must be able to identify missing, inconsistent, or illogical data before it reaches analysis. This skill ensures that trial data reflects what happened at the site, not what was assumed or incorrectly recorded. Weak data review leads to unreliable results and last-minute rework during close-out.
Resolving Data Inconsistencies Identifying issues is not enough. CDM professionals must raise precise queries, track responses, and ensure corrections are properly documented. This ability directly impacts audit readiness, because regulators expect to see not just corrected data, but a clear record of how and why changes were made.
CRF-Based Data Understanding Understanding How Case Report Forms are designed and used helps ensure that patient data is captured consistently across sites. Poor CRF understanding often results in incorrect or incomplete data entry, increasing query volume, and slowing down the entire trial.
CDM Process and Regulatory Skills
Clinical Trial Flow Awareness CDM professionals must understand how data moves across study start-up, conduct, and close-out phases. This awareness helps them prioritize reviews, anticipate risks, and prevent bottlenecks at critical milestones such as database lock and submission preparation.
Protocol and Data Management Plan Interpretation The protocol and Data Management Plan define what data must be collected and how it should be handled. CDM professionals must be able to interpret these documents accurately, because misalignment between protocol intent and data handling rules leads to compliance issues and regulatory questions.
Regulatory Readiness Awareness CDM teams must work with the assumption that trial data will be inspected. Understanding audit trails, traceability, and inspection expectations ensures that data remains defensible throughout the study and reduces the risk of findings during regulatory review. These controls support ICH GCP compliance.
Tools and Systems Skills
EDC Platform Proficiency Hands-on experience with Electronic Data Capture systems is essential for managing CRFs, queries, validations, and database lock activities. Proficiency in EDC platforms enables CDM teams to control data quality efficiently and respond quickly to issues as they arise.
Clinical Coding Standards Knowledge of coding dictionaries such as MedDRA and WHO-DD allows CDM professionals to standardize adverse events and medication data. Consistent coding is critical for accurate safety analysis and regulatory reporting.
Reporting and Review Tools Dashboards and reports are used to track data completeness, open queries, validation status, and site performance. The ability to interpret these reports helps CDM teams identify risks early and take corrective action before issues escalate.
Key Deliverables of Clinical Data Management
In clinical research, data only has value when it is complete, traceable, and acceptable for regulatory review. Clinical Data Management is measured not by how much data is collected, but by the quality and usability of what is ultimately delivered.
Clinical Data Management delivers three critical outcomes:
Clean and complete datasets that reflect real patient outcomes
Analysis-ready, locked databases for reporting and submission
Regulatory-compliant datasets and documentation required for review by authorities
1. Clean and Complete Clinical Trial Data
What this means Clinical data management delivers datasets where patient information is accurate, consistent, and complete across all trial sites and data sources. Data issues are identified early, corrected systematically, and fully documented throughout the study lifecycle.
Why this matters If data is incomplete or inconsistent, trial results cannot be trusted. Clean data ensures that analyses reflect real patient outcomes and prevents last-minute rework that can delay database lock or raise regulatory concerns.
2. Analysis-Ready and Locked Database
What this means CDM teams deliver a finalized database in which all queries are resolved, safety data is reconciled, and validation checks are complete. Once the database is locked, no further changes are permitted.
Why this matters The locked database becomes the single source of truth for statistical analysis and clinical study reports. Any unresolved issues at this stage directly affect analysis of timelines and can delay regulatory submissions.
3. Regulatory-Compliant Datasets and Documentation
What this means Clinical data management produces standardized, traceable datasets supported by complete documentation, including audit trails and validation reports. These deliverables demonstrate how data was collected, reviewed, and finalized.
Why this matters Regulatory authorities assess not just study results, but the integrity of the data behind them. Agencies such as the U.S. FDA require submitted study data to follow defined data standards for review by CDER and CBER. Without regulatory-ready datasets and documentation, even well-designed trials face delays, additional scrutiny, or rejection.
The Road Ahead
Clinical Data Management sits at the core of how modern clinical trials succeed or fail. It determines whether trial data is reliable, and acceptable for regulatory review. From study planning to database lock, CDM connects patient data with scientific analysis and regulatory decision-making, directly influencing trial timelines, data integrity, and patient safety.
As clinical trials become more global, data-driven, and inspection-focused, the demand for professionals who understand real-world data processes continues to grow. Building a career in clinical data management requires more than theoretical knowledge; it requires hands-on exposure to how data is handled across a trial lifecycle. Programs like the Advanced Diploma in Clinical Research at Clinical Research Training Institute focus on this practical understanding, preparing learners to step into clinical data roles with clarity and industry relevance.
Frequently Asked Questions – (FAQs)
1. What issues does clinical data management help avoid clinical trials?
Clinical data management prevents issues such as inconsistent data entry, unresolved discrepancies, misaligned safety reporting, and missing documentation, all of which can delay database lock and regulatory review.
2. How does a Data Management Plan guide for a clinical trial?
A Data Management Plan defines how data will be collected, reviewed, validated, and locked. A weak DMP leads to inconsistent handling of data across sites, while a clear DMP reduces rework and inspection risk.
3. Why are data queries important in clinical trials?
Query management is the process of identifying data issues, raising questions to sites, and tracking responses. Poor query management causes unresolved discrepancies to pile up, delay data cleaning, and database locking.
4. How do CRFs affect data accuracy in clinical studies?
A Case Report Form determines how patient data is captured at sites. Poorly designed CRFs increase data entry errors and query volume, directly affecting clinical trial data quality.
5. How does Electronic Data Capture support the clinical data management process?
Electronic Data Capture systems standardize data collection, apply real-time data validation checks, and maintain audit trails, helping CDM teams manage data efficiently across multiple trial sites.
6. Why is database lock considered a critical milestone?
Database lock marks the point at which data is finalized, and no further changes are allowed. Any unresolved issues at this stage directly impact analysis timelines and regulatory submissions.
7. How does clinical data management address data privacy and security?
Clinical data management systems control user access, track all data changes, and protect patient identifiers, ensuring data privacy and security throughout the clinical data management lifecycle.
8. Why is medical coding essential for patient safety data?
Medical coding standardizes adverse events and medication data, allowing consistent safety analysis and supporting regulatory review across different sites and regions.
9. What is the role of audit trails in ICH GCP compliance?
Audit trails record who made data changes, when they were made, and why. Regulators rely on audit trails to assess data integrity and verify compliance with ICH GCP guidelines.
10. How do data validation checks and source data verification work together?
Data validation checks identify inconsistencies within the database, while source data verification confirms accuracy against original patient records. Together, they support reliable data cleaning in clinical trials.
Predictive modelling in healthcare is about using patient data to make better decisions before health problems become serious. Instead of waiting for a patient’s condition to worsen, hospitals and doctors use data from past cases to understand what might happen next.
In healthcare, many problems do not appear suddenly. Patients often show small warning signs long before complications, readmissions, or emergencies occur. These signs are easy to miss when care teams are busy or working with limited information.
Predictive modeling helps identify these risks early. It supports healthcare teams in deciding who may need closer attention, extra follow-up, or timely treatment. Before looking at how it works or the methods behind it, it is important to first understand what predictive modeling means in a healthcare setting and why it is used.
In this blog, you’ll learn what predictive modeling means in a healthcare context, the kinds of problems it solves, how it works at a high level, and where it is used in real-world patient care.
What Is Predictive Modeling in Healthcare?
Predictive modeling in healthcare is the use of data to estimate what is likely to happen next, so healthcare teams can act earlier and make better decisions.
At its core, it works by looking at patterns from the past and applying them to current situations. When similar conditions appear again, predictive modeling helps signal possible risks, outcomes, or needs before they become obvious problems.
What Data Does It Use?
Predictive modeling in healthcare can use several kinds of data, depending on the problem being addressed:
Patient data for clinical data analysis, such as medical history, lab results, vital signs, medications, and treatment timelines
Operational data, such as bed availability, staff workload, length of stay, and patient flow
Administrative and claims data, including billing records, procedure codes, and healthcare utilization patterns
Population and public health data for population health analytics, such as disease trends, geographic patterns, and seasonal outbreaks
Each type of data helps predict different kinds of outcomes. Patient data supports clinical care decisions, while operational and population data support hospital planning and public health management.
POST GRADUATE DIPLOMA IN
AI and ML in Healthcare
Acquire the skills to develop and apply Artificial Intelligence and Machine Learning algorithms to medical diagnosis, treatment planning, and drug discovery. This program focuses on transforming healthcare delivery through predictive analytics and automated data-driven insights.
What Problems Predictive Modeling Solves in Healthcare
Predictive modeling is used in healthcare because many important decisions must be made early, often before problems are obvious. In real clinical environments, doctors and care teams work under time pressure and with incomplete information. When risks are identified late, patients face avoidable complications, and healthcare systems absorb unnecessary strain. Predictive modeling exists to reduce this gap by helping teams anticipate what may happen next and act while there is still time to intervene. Below we are discussing some of the most common predictive modeling healthcare applications today.
Will This Patient Deteriorate?
In hospitals, patient deterioration is rarely sudden. Most patients show subtle warning signs long before a serious event occurs. Small changes in vital signs, lab values, oxygen levels, or mental status may indicate that a patient’s condition is worsening. However, these changes are easy to miss during routine checks, especially when clinicians are responsible for many patients at once.
Predictive modeling helps by analyzing patterns across time rather than isolated measurements. By comparing current patient trends with historical cases, it can flag patients who are at higher risk of deterioration even when they appear clinically stable. This allows care teams to increase monitoring, adjust treatment, or escalate care earlier, reducing the chances of sudden emergencies such as cardiac arrest or unplanned ICU transfer.
Case Study: Early Warning Systems for Patient Deterioration – Acute Care & ICU Settings (Philips, 2020)
Hospitals have implemented predictive early warning systems that continuously analyze vital signs and monitoring data to detect patient deterioration hours before visible clinical collapse.
These systems generate risk scores that alert care teams when subtle physiological patterns suggest rising danger, even if patients appear clinically stable during routine checks.
In real-world deployments, hospitals reported up to a 35% reduction in adverse events and an over 86% decrease in cardiac arrests after integrating predictive alerts into clinical workflows.
This case demonstrates how predictive modeling significantly improves patient safety by enabling earlier intervention and faster escalation of care.
Will This Patient Be Readmitted?
Hospital readmissions are often driven by issues that occur after discharge rather than during the hospital stay itself. Patients may struggle with medication management, fail to attend follow-up appointments, misunderstand discharge instructions, or lack adequate support at home. These factors are difficult for clinicians to assess consistently using manual judgment alone.
Predictive modeling helps identify patients who are more likely to be readmitted before they leave the hospital. By recognizing patterns associated with past readmissions, healthcare teams can focus additional support on higher-risk patients. This may include clearer discharge education, early follow-up appointments, medication reconciliation, or post-discharge check-ins. The goal is not to prevent discharge, but to improve recovery and reduce avoidable returns to the hospital.
Case Study: Reducing Hospital Readmissions with Predictive Modeling – Corewell Health (USA)
Corewell Health used predictive modeling to identify patients at high risk of 30-day hospital readmission at the time of discharge. The system combined clinical data with behavioral and social factors to generate risk scores, which were reviewed by clinicians and care coordination teams.
Rather than relying on prediction alone, high-risk patients received targeted follow-up support, improved discharge planning, and focused transition-of-care interventions. This approach demonstrates direct, real-world use of predictive analytics to improve healthcare outcomes.
Over approximately 20 months, this strategy prevented around 200 avoidable readmissions and generated nearly USD 5 million in cost savings.
This case highlights that predictive modeling in healthcare works best when risk identification is paired with human clinical judgment and timely clinical action, rather than fully automated decisions.
Who Needs Urgent Attention Right Now?
In emergency departments and busy hospital wards, not all patients can be treated immediately. Patients often arrive with similar symptoms, and some may appear stable even though their condition is likely to worsen in the next few hours. Relying only on visible symptoms or arrival order can delay care for those at highest risk.
Predictive modeling supports prioritization by estimating which patients are more likely to deteriorate in the near term. This allows care teams to direct attention toward higher-risk patients sooner, even if they do not yet appear critically ill. As a result, urgent cases are less likely to be overlooked, and delays that lead to adverse outcomes can be reduced.
Case Study: Suicide Risk Prediction Using EHR Data – Vanderbilt University Medical Center (USA)
Vanderbilt University Medical Center developed a machine learning–based system that analyzes routine electronic health record (EHR) data to estimate suicide risk during patient encounters. The model runs silently in the background, grouping patients by risk level so clinicians can identify individuals who may need mental health screening even when no obvious warning signs are present.
During evaluation, the system identified a high-risk group that accounted for over one-third of subsequent suicide attempts, demonstrating how predictive modeling can surface hidden risk early.
This case highlights how predictive analytics supports earlier screening and prevention for rare but critical outcomes by complementing clinical judgment rather than replacing it.
Will This Patient Be Readmitted?
Hospital readmissions are often driven by issues that occur after discharge rather than during the hospital stay itself. Patients may struggle with medication management, fail to attend follow-up appointments, misunderstand discharge instructions, or lack adequate support at home. These factors are difficult for clinicians to assess consistently using manual judgment alone.
Predictive modeling helps identify patients who are more likely to be readmitted before they leave the hospital. By recognizing patterns associated with past readmissions, healthcare teams can focus additional support on higher-risk patients. This may include clearer discharge education, early follow-up appointments, medication reconciliation, or post-discharge check-ins. The goal is not to prevent discharge, but to improve recovery and reduce avoidable returns to the hospital.
Case Study: Reducing Hospital Readmissions with Predictive Modeling – Corewell Health (USA)
Corewell Health used predictive modeling to identify patients at high risk of 30-day hospital readmission at the time of discharge. The system combined clinical data with behavioral and social factors to generate risk scores, which were reviewed by clinicians and care coordination teams.
Rather than relying on prediction alone, high-risk patients received targeted follow-up support, improved discharge planning, and focused transition-of-care interventions. This represents a direct and practical use of predictive analytics for improving healthcare outcomes.
Over approximately 20 months, this approach prevented around 200 avoidable readmissions and generated nearly USD 5 million in cost savings.
Who Needs Urgent Attention Right Now?
In emergency departments and busy hospital wards, not all patients can be treated immediately. Patients often arrive with similar symptoms, and some may appear stable even though their condition is likely to worsen in the next few hours. Relying only on visible symptoms or arrival order can delay care for those at highest risk.
Predictive modeling supports prioritization by estimating which patients are more likely to deteriorate in the near term. This allows care teams to direct attention toward higher-risk patients sooner, even if they do not yet appear critically ill. As a result, urgent cases are less likely to be overlooked, and delays that lead to adverse outcomes can be reduced.
Case Study: Suicide Risk Prediction Using EHR Data – Vanderbilt University Medical Center (USA)
Vanderbilt University Medical Center developed a machine learning–based system that analyzes routine electronic health record (EHR) data to estimate suicide risk during patient encounters.
The model runs silently in the background, grouping patients by risk level so clinicians can identify individuals who may need mental health screening even when no obvious warning signs are present.
During evaluation, the system identified a high-risk group that accounted for over one-third of subsequent suicide attempts, demonstrating how predictive modeling can surface hidden risk early.
This case highlights how predictive analytics enables earlier screening and prevention for rare but critical outcomes by supporting proactive clinical decision-making.
Which Patients Need Follow-Up Care?
After diagnosis or treatment, maintaining follow-up is a major challenge in healthcare. Some patients miss appointments, delay recommended tests, or discontinue treatment, which can lead to late diagnoses, disease progression, or emergency visits. Following up with every patient at the same level is not realistic given limited resources.
Predictive modeling helps identify patients who are more likely to miss follow-ups or develop complications if care is interrupted. By focusing reminders, outreach, and follow-up efforts on these patients, healthcare teams can improve continuity of care and prevent avoidable deterioration outside the hospital setting.
Case Study: Early Sepsis Detection Using Machine Learning – ICU & Hospital Research Deployments
Machine learning models trained on large clinical datasets have demonstrated the ability to identify sepsis risk earlier than traditional scoring systems.
By continuously analyzing trends in vital signs and laboratory results, these models detect early warning signals hours before sepsis becomes clinically obvious.
Research-based deployments consistently show earlier detection and improved risk discrimination, forming the foundation for real-time sepsis alert systems now used in hospitals.
This case reinforces the role of predictive modeling in preventing life-threatening complications through timely identification and early clinical intervention.
How Predictive Modeling Works in Healthcare (Explained Through Real Healthcare Scenarios)
Predictive modeling in healthcare is not built in isolation by data teams. It is shaped by real clinical problems, real patient behavior, and real operational constraints. Each step in the process exists because healthcare decisions carry risk, and getting even one step wrong can lead to unsafe or misleading predictions.
To understand how predictive modeling works in practice, it helps to walk through the process as it unfolds inside a healthcare setting.
Defining the Problem: Starting With a Care Gap
Predictive modeling begins when healthcare teams notice a recurring problem they cannot reliably manage using observation alone. For example, a hospital may realize that many patients who end up in the ICU showed warning signs earlier, but those signs were not recognized in time. In another case, leadership may notice that readmissions are high even though discharge criteria are being followed correctly.
At this stage, the goal is not to build a model, but to clearly define what needs to be predicted. Is the priority to identify deterioration early? To prevent readmissions? To prioritize patients during peak workload? In healthcare, vague questions lead to unsafe predictions, so this step ensures the model is built to support a specific clinical decision.
Gathering Data: Reconstructing the Patient Journey
Once the problem is clear, healthcare teams collect data that reflects how care actually unfolds. For patient deterioration, this includes vital signs, lab results, oxygen levels, medications, and how these values change over time. For readmissions, the data may include discharge timing, medication changes, prior hospital visits, and follow-up history.
This step is critical because healthcare outcomes are rarely caused by a single factor. Predictive modeling depends on understanding patterns across entire patient journeys, not isolated snapshots. Without the right data, predictions may overlook the very signals clinicians are trying to catch early.
Preparing the Data: Making Sure the Story Is Accurate
Healthcare data is often messy because it is collected across multiple systems and departments. A patient’s lab results may be recorded in one system, vital signs in another, and discharge information elsewhere. Incomplete or inconsistent records can distort patterns and create false signals.
Before any learning can occur, the data must be aligned so it tells a consistent story. This step matters because predictive models do not understand context; they only learn from what they are given. In healthcare, poor data preparation can translate directly into unsafe recommendations.
Learning From Past Outcomes: Finding What Actually Comes Before Trouble
With reliable data in place, predictive modeling looks backward before it looks forward. It examines previous patient cases to understand what typically happened before certain outcomes occurred. For example, it may reveal that patients who were later readmitted often showed specific lab trends, medication changes, or follow-up gaps in the days before discharge.
This step is important because healthcare intuition alone is not enough at scale. While clinicians may recognize patterns in individual cases, predictive modeling helps confirm which signals consistently matter across hundreds or thousands of patients.
Testing Predictions: Making Sure Patterns Hold Up
Not every pattern discovered in data is meaningful. Some patterns appear by chance or reflect temporary conditions. Before predictions are trusted, they must be tested against real historical cases to ensure they reliably identify risk without generating excessive false alarms.
In healthcare, this step is essential for safety. A model that flags too many patients creates alert fatigue, while one that misses risk undermines trust. Testing ensures predictions strike the right balance between sensitivity and usefulness in real clinical environments.
Integrating Into Care: Making Predictions Usable
Even accurate predictions are useless if they do not fit into clinical workflows. Healthcare professionals do not have time to interpret complex outputs or separate dashboards. Predictive insights must be presented in a simple, actionable form, such as a risk score or early warning indicator within existing systems.
This step matters because healthcare decisions are made quickly and often under pressure. Predictive modeling succeeds only when it supports, rather than disrupts, how care is delivered.
Acting With Clinical Judgment: Supporting, Not Replacing, Care Teams
Predictions are designed to draw attention, not dictate action. When a patient is flagged as high risk, clinicians assess the situation in context, considering factors that data may not capture, such as patient behavior, social support, or recent changes in condition.
This step exists because healthcare is not deterministic. Predictive modeling provides early signals, but human judgment remains essential to ensure safe and appropriate care.
Monitoring Over Time: Keeping Predictions Relevant
Healthcare does not stand still. Treatment protocols evolve, patient populations change, and hospital workflows adapt. Predictive models must be monitored to ensure they remain accurate and fair as conditions change.
This step is especially important in healthcare because outdated predictions can be as harmful as incorrect ones. Ongoing monitoring ensures that predictive modeling continues to support patient safety and care quality over time.
POST GRADUATE DIPLOMA IN
Biostatistics
Master the development and application of statistical methods to health data. This program equips you with the technical expertise to analyze complex biomedical data, interpret research findings, and drive evidence-based practice in public health and life sciences.
Advanced Biostatistical Modeling, Survival Analytics, Epidemiological Data Analysis, Data Visualization, and Proficiency in Statistical Software (R/SAS)
How Healthcare Systems Learn Patterns: Algorithms Used in Predictive Modeling
Once healthcare data is prepared and past outcomes are understood, the next question is simple: how does the system actually learn from this information? This is where algorithms come in.
An algorithm, in this context, is a method that helps the system learn patterns from past healthcare data. When an algorithm is trained on real data, it produces a predictive model that can estimate risk for new patients or situations. Different healthcare problems require different learning approaches, which is why multiple algorithms are used instead of one universal method.
Many healthcare decisions involve a clear yes-or-no question. Will a patient be readmitted? Is there a high risk of complications? Should closer monitoring be triggered? Logistic regression is commonly used in these situations because it focuses on estimating probability rather than making absolute claims.
Healthcare teams value this approach because it produces clear risk scores and is relatively easy to interpret. Clinicians can understand which factors contribute to higher risk, making it suitable for decisions that must be explained, reviewed, or audited. It is often used as a first-line approach for clinical risk prediction because it balances simplicity, transparency, and usefulness.
Decision Trees: Mimicking Clinical Reasoning
In many healthcare settings, decisions follow logical steps. Clinicians often think in terms of conditions and thresholds, such as whether a lab value is above or below a certain level or whether specific symptoms are present. Decision trees reflect this type of reasoning by breaking decisions into a sequence of simple rules.
This approach is useful when explainability is critical. Clinicians can follow the decision path and understand how a conclusion was reached. While decision trees may not always provide the highest accuracy, they align well with clinical workflows and guideline-based decision-making.
Random Forests: Improving Reliability in Complex Cases
Healthcare data is rarely clean or consistent. A single decision tree can be sensitive to small variations in data, which may lead to unstable predictions. Random forests address this by combining many decision trees and using their collective output to make predictions.
This approach improves reliability and accuracy, especially when dealing with complex patient data from electronic health records. While random forests are harder to explain than a single decision tree, they are often used when healthcare teams need stronger performance and are willing to trade some interpretability for better prediction quality.
Neural Networks and Deep Learning: Learning From Complex Data
Some healthcare data is too complex for rule-based or statistical approaches. Medical images, physiological signals, and genomic data contain patterns that are difficult for humans to define explicitly. Neural networks and deep learning are designed to learn these patterns directly from large volumes of data.
These approaches are commonly used in areas such as medical imaging and diagnostics, where accuracy is critical and patterns are not obvious. Because they are harder to interpret, they are usually deployed with additional validation and oversight, especially in clinical environments.
Survival Analysis: Understanding When Events May Happen
In healthcare, timing often matters as much as risk. Clinicians may need to know not just whether an event will occur, but when it is likely to occur. Survival analysis focuses on time-based outcomes, such as how long before a patient is readmitted or how disease risk changes over time.
This approach is widely used in clinical research and long-term care planning because it handles follow-up data naturally and provides insight into how risk evolves. It is particularly valuable when outcomes unfold gradually rather than immediately.
Why Healthcare Uses Multiple Algorithms
No single algorithm can handle every healthcare problem safely or effectively. Some situations demand clarity and explainability, while others demand higher accuracy or the ability to handle complex data. Healthcare teams choose algorithms based on the clinical question, the type of data available, and how predictions will be used in practice.
This is why predictive modeling in healthcare is not about finding the “best” algorithm, but about choosing the right learning approach for the right decision.
Limitations of Predictive Modeling in Healthcare
Predictive modeling can improve decision-making in healthcare, but it is not a flawless solution. Because predictions influence real patient care, understanding the limitations of predictive modeling is essential. When these systems are misunderstood or overtrusted, they can introduce new risks rather than reduce existing ones.
Data Quality Directly Limits Prediction Quality
Predictive models depend entirely on the data they learn from. In healthcare, data is often incomplete, inconsistent, or fragmented across multiple systems. Patients may receive care from different hospitals, labs, and providers, and important information may be missing or recorded differently. When predictive models are trained on this kind of data, they learn from an imperfect representation of reality. This can result in predictions that appear precise but are fundamentally unreliable. Predictive modeling cannot correct poor data quality; it only reflects it.
Bias in Historical Data Can Be Reinforced
Predictive modeling learns from past healthcare decisions and outcomes. If historical data reflects unequal access to care, delayed treatment, or systemic bias against certain patient groups, those patterns can be unintentionally carried forward. This can lead to underestimating risk for some populations while overestimating it for others. In healthcare, where equity and safety are critical, unmanaged bias can worsen existing disparities rather than improve care.
Limited Understanding of Human and Social Context
Predictive models do not understand patients as individuals. They lack awareness of social circumstances, emotional state, family support, or sudden life changes unless these factors are explicitly captured in data. A patient may be classified as low risk based on clinical indicators while still facing significant challenges outside the healthcare system. This limitation makes it essential for predictions to be interpreted alongside clinical judgment and real-world context.
Risk of Over-Reliance and Alert Fatigue
Predictive modeling produces probabilities, not certainties. However, in practice, there is a risk of treating predictions as definitive answers. Over-reliance on risk scores or alerts can lead to unnecessary interventions or missed edge cases. In busy clinical environments, frequent alerts can also cause fatigue, reducing attention to genuinely critical signals. Predictive modeling should guide attention, not replace decision-making.
Models Can Become Outdated Over Time
Healthcare environments evolve continuously. Treatment protocols change, patient populations shift, and new conditions emerge. Predictive models trained on older data may lose accuracy if they are not regularly reviewed and updated. Without ongoing monitoring, even well-performing models can become misleading, creating false confidence in outdated predictions.
Regulatory, Ethical, and Trust Constraints
Healthcare is a highly regulated domain. Predictive models must be explainable, auditable, ethical, and aligned with patient safety standards. Clinicians are less likely to trust systems they cannot understand or challenge. Patients may also feel uneasy when care decisions appear to be driven by opaque systems. These concerns limit how predictive modeling can be deployed, especially in high-stakes clinical settings.
Why These Limitations Matter
Predictive modeling delivers value only when its limitations are clearly understood. It is most effective when used as a decision-support tool, not a decision-maker. Recognizing where predictive modeling can fail helps healthcare teams apply it responsibly, combine it with clinical expertise, and avoid false confidence. In healthcare, the goal is not perfect prediction, but safer and earlier decision-making.
Future Trends in Predictive Modeling in Healthcare
Predictive modeling in healthcare is evolving, not because of flashy algorithms, but because healthcare itself is changing. Data availability is improving, care is moving beyond hospital walls, and expectations around safety and accountability are rising. These shifts are shaping how predictive modeling will be built and used in the coming years.
From Static Predictions to Real-Time Risk Monitoring
Early predictive models were often run periodically, using snapshots of patient data. The future is moving toward continuous, real-time risk assessment. Instead of generating a score once a day or at discharge, predictive systems will update risk levels as new lab results, vitals, or monitoring data arrive.
This matters because patient conditions change quickly. Real-time prediction allows healthcare teams to respond to early signals as they emerge, rather than discovering risk after deterioration has already begun.
Predictive Modeling Extending Beyond Hospitals
Predictive modeling is no longer confined to inpatient care. As healthcare shifts toward outpatient, home-based, and virtual care, predictive systems are being used to monitor patients outside traditional clinical settings. Data from wearables, remote monitoring devices, and follow-up interactions are increasingly incorporated into risk assessment.
This expansion supports earlier intervention for chronic conditions, post-discharge recovery, and home-based care, helping prevent avoidable hospital visits before they occur.
Greater Emphasis on Explainability and Transparency
As predictive modeling becomes more embedded in care decisions, explainability is becoming non-negotiable. Clinicians need to understand why a patient is flagged as high risk, not just that they are. Regulators and healthcare organizations are also demanding clearer documentation of how predictions are generated and used.
Future predictive systems will place greater emphasis on transparent reasoning, traceable inputs, and interpretable outputs so predictions can be reviewed, questioned, and trusted.
Increased Use of Combined and Hybrid Approaches
Healthcare problems rarely fit neatly into one modeling approach. Future predictive systems will increasingly combine multiple learning methods to balance accuracy, timing, and interpretability. Simpler approaches may be used for early screening, while more complex methods refine predictions in the background.
This hybrid approach reflects a practical shift away from searching for a single “best” model toward building systems that work reliably across different stages of care.
Stronger Governance, Monitoring, and Accountability
As predictive modeling influences more clinical decisions, healthcare organizations are placing stronger controls around how models are deployed and maintained. Continuous monitoring for accuracy, bias, and unintended consequences is becoming standard practice rather than an afterthought.
This trend reflects a broader understanding that predictive modeling is not a one-time implementation, but a living system that must be governed throughout its lifecycle.
Beyond individual patient care, predictive modeling is increasingly used to support population health and system-level planning. Health systems and public health agencies are using predictions to anticipate demand, manage staffing, prepare for disease surges, and allocate resources more effectively.
At this level, predictive modeling helps healthcare systems prepare rather than react, improving resilience during periods of stress.
Conclusion
Predictive modeling is not about predicting the future with certainty. In healthcare, its value lies in helping teams recognize risk earlier, make more informed decisions, and intervene before problems escalate. When used responsibly, it supports safer care, better prioritization, and more efficient use of limited resources.
As healthcare continues to generate more data and operate under increasing pressure, the ability to interpret patterns and act early will only become more important. Predictive modeling provides a structured way to do that, but its impact depends on how well it is understood, implemented, and combined with clinical judgment.
For professionals looking to build practical skills in this space, understanding predictive modeling in a healthcare context is no longer optional. Clinical Research Training Institute offers industry-ready programs, including AI and ML in Healthcare, designed to bridge the gap between healthcare knowledge and real-world data applications. These programs focus on applied learning that aligns with how predictive modeling is actually used across hospitals, clinical research, and digital health.
FAQs
1. What Is the Role of Predictive Analytics in Healthcare?
Predictive analytics helps healthcare teams make better decisions ahead of time. Instead of reacting after something goes wrong, it helps identify risks early, prioritize patients who need attention, and improve planning for care and resources.
2. How Are Machine Learning and Data Science Used in Healthcare?
Machine learning and data science are used to analyze large amounts of healthcare data, such as patient records, test results, and medical images. They help find patterns that are hard to see manually and support predictions related to diagnosis, risk, and treatment outcomes.
3. How Is Predictive Modeling Used in Medical Research?
In medical research, predictive modeling helps researchers understand how diseases progress and how patients may respond to treatments. It is used to study trends, identify risk factors, and support better study design and clinical decision-making.
4. What Are the Benefits of Predictive Analytics for Healthcare Outcomes?
Predictive analytics improves healthcare outcomes by enabling early intervention, reducing avoidable complications, lowering readmission rates, and supporting more personalized care. It also helps healthcare systems work more efficiently.
5. How Is Predictive Modeling Used in Healthcare?
Predictive modeling is used to identify high-risk patients, support clinical decisions, prioritize care, plan follow-ups, and reduce preventable hospital visits. It helps healthcare teams focus their efforts where they matter most.
6. What Are Examples of Predictive Analytics in Healthcare?
Common examples include predicting patient deterioration, identifying readmission risk, detecting disease early, prioritizing emergency care, and forecasting long-term health outcomes for chronic conditions.
7. How Does Machine Learning Improve Healthcare Predictions?
Machine learning improves healthcare predictions by learning from large volumes of past data and continuously refining patterns. This allows predictions to become more accurate over time, especially in complex cases where simple rules are not enough.
Functional Genomics in Healthcare
Functional genomics in healthcare exists because knowing what is written in DNA is no longer enough to understand how diseases behave in real patients. People with the same diagnosis and similar genetic reports often experience very different symptoms, disease progression, and treatment outcomes.
Traditional genetic testing identifies DNA variations, but it often cannot explain how those genes behave inside the body or why outcomes vary so widely. Clinical resources such as MedlinePlus from the U.S. National Library of Medicine highlight this limitation, which pushed healthcare toward approaches that study gene activity, biological pathways, and molecular behavior instead of DNA sequence alone.
This blog introduces what functional genomics is, why it became necessary in modern healthcare, and how it is applied today through real-world examples.
Functional genomics in healthcare studies how genes and their products (RNA and proteins) function in the body to explain disease behavior and treatment response. Instead of focusing only on DNA sequences, it analyzes gene expression, molecular pathways, and biological activity to improve diagnosis, therapy selection, and understanding of disease progression.
What Is Functional Genomics?
Functional genomics is the study of how genes function inside the body, not just what their DNA sequence looks like. Instead of focusing only on which genes are present, it examines how genes behave, when they are active, and how they influence biological processes in real conditions.
Genes are constantly being switched ON and OFF. Some activate only during illness or stress, while others remain silent. Functional genomics focuses on gene expression analysis to understand what is actually happening inside cells as diseases develop or respond to treatment.
Traditional Genomics vs Functional Genomics
Traditional genomics provides static information about DNA, which rarely changes. Functional genomics captures dynamic biological activity, showing how genes interact with proteins, pathways, and cellular systems over time.
In simple terms, traditional genomics tells us what could happen, while functional genomics explains what is happening right now. This distinction is critical for understanding complex diseases.
Case Study: Rheumatoid Arthritis and the Limits of Genetic Risk
Aspect
Details
The Problem
Thousands of genetic variants are linked to rheumatoid arthritis, yet many patients do not respond to treatment.
What Functional Genomics Revealed
Analysis of DNA folding and gene regulation in immune cells identified which genes were actually affected.
What Changed
New disease-driving pathways were identified that required activation rather than inhibition.
Why This Matters
Functional genomics transformed genetic associations into actionable biology.
What Functional Genomics Is NOT
Not just DNA sequencing: It focuses on gene activity, not only DNA reading.
Not a single test: It combines multiple biological datasets.
Not limited to cancer: Used in autoimmune, neurological, metabolic, and rare diseases.
Not a replacement for clinicians: It supports medical decisions, not replaces them.
Not only experimental: Already used in diagnostics and treatment planning.
What Is the Need for Functional Genomics?
Modern healthcare reached a point where genetic information alone stopped being sufficient. DNA sequencing can identify mutations, but it often fails to explain disease behavior or treatment outcomes.
Diseases such as cancer and autoimmune disorders involve networks of genes and pathways that change over time. Functional genomics addresses this by focusing on gene activity rather than genetic potential.
Functional genomics is actively used in pharmaceutical research. Companies like AstraZeneca apply it to link genetic data with biological function and improve drug discovery outcomes.
Case Study: Genome-Scale Cancer Target Discovery
Aspect
Details
The Problem
Cancer drug development fails frequently and late in trials.
What Functional Genomics Revealed
CRISPR screening identified genes cancers truly depend on.
What Changed
Drug targets could be prioritized with higher success probability.
Why This Matters
Functional genomics reduces wasted effort in drug discovery.
Professional Diploma in
Bioinformatics and Metabolomics
Build real-world skills in bioinformatics and metabolomics used across healthcare, pharma, and life sciences. Learn to analyze multi-omics data, identify biomarkers, and translate complex biological data into actionable insights using modern analytical pipelines.
Functional genomics is applied wherever understanding biological activity matters more than simply knowing which genes exist. In healthcare, this shift has transformed how diseases are detected, classified, treated, and studied over time.
Early Disease Detection
Many diseases begin with molecular changes long before symptoms appear. Functional genomics helps detect these early signals by identifying abnormal patterns of gene activity and pathway disruption.
In cancers, altered gene expression can signal tumor development before it becomes visible on scans. In neurological conditions such as Alzheimer’s or Parkinson’s disease, early disruptions in neuronal signaling and metabolic pathways can be detected years before clinical diagnosis.
By detecting abnormal gene activity before symptoms appear, functional genomics strengthens molecular diagnostics and enables earlier, more accurate intervention.
Case Study: Gastric Cancer Drug Synergy Explained by Gene Function
Aspect
Details
The Problem
Gastric cancer is highly resistant to chemotherapy, and many drug combinations fail despite targeting known pathways.
What Functional Genomics Revealed
Screening revealed that a drug combination worked because one drug blocked the cancer cell’s drug-efflux mechanism.
What Changed Because of It
Researchers understood why the combination worked and identified patients most likely to benefit.
Why This Case Matters
Functional genomics revealed what drugs were actually doing inside cells.
Personalized Treatment Planning
Patients with the same diagnosis often respond differently to the same treatment. Functional genomics explains this by revealing how active disease-related pathways are in individual patients.
In oncology, functional profiling identifies whether tumors are driven by growth signaling, immune evasion, or metabolic changes. In autoimmune diseases, it distinguishes inflammatory patterns that appear similar clinically but require different therapies.
By aligning treatment choices with biological behavior rather than labels, functional genomics reduces trial-and-error medicine.
Disease Subtyping and Risk Stratification
Many diseases that appear identical under standard testing are biologically different at the molecular level. Functional genomics enables disease subtyping based on gene activity rather than symptoms alone.
This is especially important in cancers, blood disorders, and neurological diseases, where molecular subtypes predict aggressiveness, recurrence risk, and long-term outcomes.
Drug Target Discovery and Validation
Targeting the wrong biological signal leads to failed therapies. Functional genomics helps identify which genes and pathways are actually driving disease.
By studying gene activity and pathway behavior, researchers validate whether targets play a causal role in disease progression. This improves target selection and reduces late-stage drug failures.
Understanding Rare and Undiagnosed Disorders
Rare genetic disorders often remain unexplained even after DNA sequencing. Functional genomics bridges this gap by showing how genetic changes disrupt biological pathways.
In inherited metabolic disorders, neuromuscular diseases, and rare epileptic syndromes, functional analysis clarifies whether a variant is harmful and how it affects the body.
Predicting Treatment Response and Resistance
Functional genomics predicts how patients respond to therapies by studying gene activity linked to drug metabolism, signaling pathways, and resistance mechanisms.
In cancer treatment, it explains why resistance develops and guides alternative strategies before disease progression worsens.
Case Study: Diagnosing Rare Diseases with RNA-Based Functional Genomics
Aspect
Details
The Problem
Many rare disease patients remain undiagnosed even after DNA sequencing.
What Functional Genomics Revealed
RNA sequencing showed how variants affected gene expression and splicing.
What Changed Because of It
Diagnostic accuracy improved for patients with no prior answers.
Why This Case Matters
Functional genomics turns uncertain genetic data into diagnoses.
Monitoring Disease Progression and Treatment Effectiveness
Functional genomics is used to track how diseases evolve over time. Changes in gene expression can indicate whether a disease is progressing, stabilizing, or responding to therapy.
This allows clinicians to adjust treatment plans based on biological response rather than waiting for symptoms or imaging changes.
Beyond One-Time Testing
Functional genomics is increasingly used for continuous monitoring by tracking changes in biological activity over time. This supports dynamic care decisions based on molecular response rather than delayed clinical signs.
Supporting Systems Biology and Integrated Care
Functional genomics aligns with systems biology by integrating gene activity with broader biological networks. Instead of viewing genes in isolation, it models how genes, proteins, and pathways interact within the body.
This systems-level understanding supports more comprehensive disease models and improves how complex conditions are diagnosed and managed across healthcare settings.
Conclusion
Functional genomics has reshaped how healthcare understands disease. Instead of relying only on DNA sequences, it focuses on how genes behave in real biological conditions, helping explain differences in disease progression, treatment response, and clinical outcomes.
As medicine becomes more personalized and data-driven, understanding gene activity and biological pathways is no longer optional across healthcare and life sciences. Functional genomics now sits at the core of modern diagnosis, research, and drug development.
From a career perspective, this shift has created growing demand for professionals who can interpret functional genomic data in clinical research, molecular diagnostics, bioinformatics, and precision medicine roles.
At CliniLaunch, we help learners build this foundation through healthcare and life sciences programs aligned with real-world clinical and research needs. Functional genomics is not just an emerging concept—it reflects how modern medicine now approaches disease, through function rather than sequence.
Frequently Asked Questions (FAQs)
FAQs
1. How is functional genomics used in everyday clinical decision-making?
Functional genomics informs how diseases are classified, which diagnostic tests are ordered, and how treatments are prioritized by revealing pathway activity and gene expression patterns rather than relying only on DNA variants.
It supports oncologists, neurologists, and immunologists in choosing therapies that align with a patient’s real-time disease biology instead of static genetic risk alone.
2. What role does functional genomics play in molecular diagnostics and early disease detection?
Functional genomics enhances molecular diagnostics by detecting abnormal gene expression and pathway disruptions before symptoms or imaging changes appear.
This enables earlier risk assessment, more accurate disease classification, and improved use of tools such as RNA-seq–based panels and multi-omics signatures in cancer, neurological, and autoimmune conditions.
3. How does functional genomics support drug discovery, target validation, and biomarker discovery?
In drug discovery, functional genomics confirms which genes and pathways actually drive disease, reducing the risk of pursuing non-causal targets.
It also accelerates biomarker discovery by linking gene activity patterns with treatment response, resistance, and disease progression, leading to more efficient and better-stratified clinical trials.
4. What skills and careers are emerging around functional genomics in healthcare?
Emerging roles include bioinformatics analyst, multi-omics data scientist, clinical genomics researcher, and molecular diagnostics specialist.
Professionals in these roles require skills in genomic data analysis, systems biology, biomarker discovery, and AI-enabled interpretation of high-throughput biological data to translate functional genomics into clinical and pharmaceutical decisions.
5. How are AI and data science changing functional genomics in modern healthcare?
AI and data science are used to analyze large functional genomics datasets, identify complex gene expression patterns, and predict treatment response or resistance.
These approaches integrate genomics, transcriptomics, proteomics, and metabolomics into practical tools for precision medicine, clinical research, and hospital-level decision support by automating variant interpretation, risk prediction, and drug response modeling.